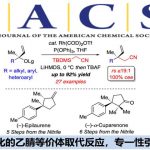
在芳香化合物中,例如对于最简单的苯环上如果能够随心所欲的在”想要的位置”直接导入”所需官能团”的话,那么作为有机化合物中最重要的结构之一,苯环衍生物的合成将变得十分简单,正因为其广泛的应用性,所以现在有很多化学家也都集中于在苯环上进行选择性官能团化的研究。
这里所说的直接,目前来讲,一般是指使用有机金属催化剂进行的芳香环上的sp2C-H键的直接官能团化。到目前为止,对于C-H键的直接官能团化取得了很大进展,比如有芳基化,烯基化,烷基化,硼化,硅化,氮化等等数不胜数的催化反应已经被报道。苯环上的直接C-H官能团化近年来就像一场飓风席卷了有机化学界。
那么,在”想要的位置”进行官能团化这个点又做得如何呢。这里主要涉及到一个位置选择性问题,也就是对芳环上的特定的C-H键进行选择性官能团反应,而这个难题到现在还没有很好的解决办法。我们都知道在考虑单取代的苯环直接官能团化的时候,必须要克服邻位、间位与对位三个位置的C-H选择性问题,因此往往在普通情况下,得到的产物都是这三者或者两者的混合物。
近年来,苯环邻位的直接C-H活化通过导向基团已经被攻克。[1, 2]
而间位・对位的研究已经进展到哪一步了呢?实际上,现在关于位置选择性的官能团化的文献报道并不多。但是,最近,东京大学的國信・金井等人,设计合成了一种新型的有机金属催化剂,实现了间位选择性芳环的C-H硼化反应。该成果被刊登在nature chemistry上。
“A meta-selective C–H borylation directed by a secondary interaction between ligand and substrate”
Kuninobu, Y.; Ida, H.; Nishi, M.; Kanai, M. Nature Chemistry 2015, 7, 712. DOI:10.1038/nchem.2322
此次,主要介绍的反应是:苯环的间位选择性官能团化以及芳香族的C-H硼化等。并且,最后我们还邀请到了本文的作者介绍整个研究的经过以及个人的感想。
间位选择性芳香C-H官能团化反应介绍
接下来就一起来看一下到目前为止报道过的一些间位选择性芳香族的C-H官能团化反应。
- 底物上取代基的立体效应
- 底物上取代基的电子效应:高价碘苯的C–H硼素化反应[3]
- 通过底物上导向基团带来的二次相互作用。
- 底物上导向基的邻位基参与:Gaunt等人的苯胺衍生物的间位选择性芳香族C-H芳香化反应。(图1)[4]
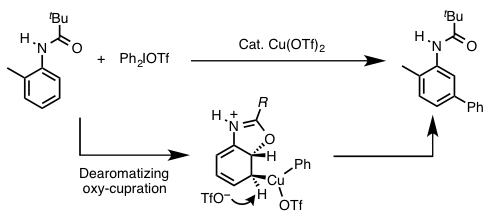
图1 邻位基团参与的间位选择性芳香族C-H芳香化反应
- 底物上导向基的导向效果:最有名的就是余金权开发的间位选择性反应了(图2)[5]
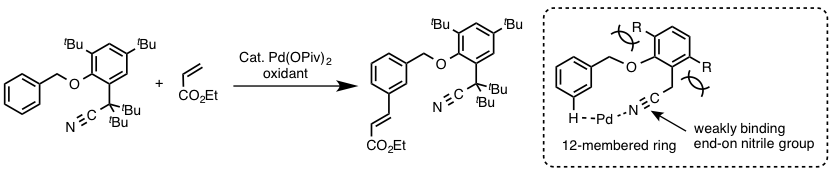
図2 利用特殊的导向基团进行的C-H烯烃化反应
上述这些反应,虽然都达到了选择性的目的,但是对于底物结构的依存性很高,这其实是由于通过催化剂控制选择性十分困难。那么我们接下来看一下芳香环的C-H键的硼化反应已经发展到什么阶段了。
芳香环C-H键的硼基化反应介绍
对于直接C-H键的硼化反应,主流是利用Ir催化剂。对于有取代的苯环的C-H硼化反应,其位置选择性来说,受底物芳香环上的取代基的立体效应影响很大。例如,下图(图3)中所示的单取代苯环作为底物进行的硼化反应,由于立体位阻,通常得不到邻位硼化,该反应最终生成2:1的间位・对位硼化体混合物[6]。另外,碘苯作为底物出现时,由于碘的电子效应,间位选择性产物会略微增加[3]。
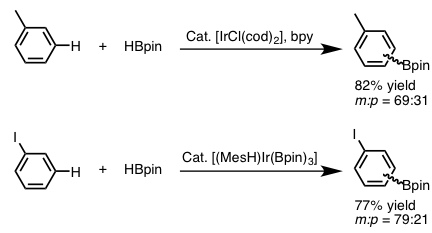
图3 碘苯的芳香C-H硼基化反应
那么如果要实现邻位的硼化反应的话,到底该怎么办呢?澤村研究室利用硅胶负载的紧凑型富电子膦配体与Ir催化剂以1:1的形式混合,形成螯合物。并且利用底物的导向基团实现了位置选择性的硼基化反应(图4)[8]。
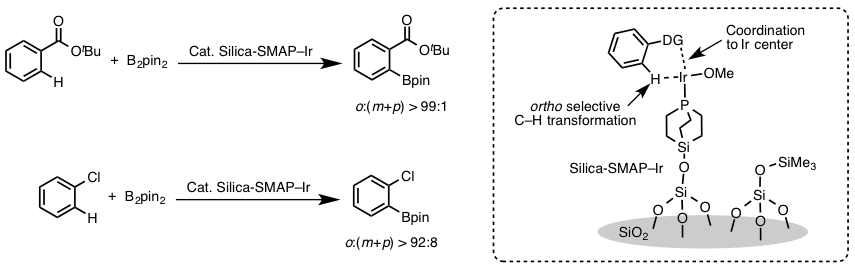
图4 利用Silica-SMAP进行的邻位选择性芳香族C-H硼基化反应
那么,对位选择性如何实现?
对位的选择性其实是最近才被伊丹组实现,他们利用立体位阻很打的二齿型磷配体与底物的立体位阻效应实现了对位选择性硼基化,其实只有实现了9成,还有1成的间位产物生成(图5)[9],可见其难度之大。

图5 高位阻磷配体控制的对位C-H硼基化反应
综上所述,利用催化剂设计实现了邻位,对位的选择性硼基化反应,最后就剩下间位的反应了。如何才能利用催化剂控制间位选择性呢?那当然还是需要能够设计出一款标配的催化剂咯!
间位选择性C-H键的直接官能团化催化剂的设计
國信・金井等人刚开始就打算以芳香酰胺或者酯作为底物,设计出一种配体恰好能够利用氢键进行识别定位,而另一侧与Ir连接的地方恰好又拉长到苯环的间位C-H位点,这样不就能够实现间位的C-H活化,进而进行硼基化反应了么?具体的他们利用「尿素骨架」作为识别基团,与底物中的羰基形成适当强度的氢键来固定住底物的方向。另外,配体的另一侧利用「4,4-双吡啶骨架」作为Ir金属的配体基团,而整个配体的长度正好把Ir活性部位延生到底物苯环中的间位C-H键处,完成了这一选择性反应。而且作者在设计底物跟催化剂时也考虑到了Ir催化的芳香族C-H硼基化反应的话,对于缺电子的芳香环来说反应是较容易进行的,所以在尿素与羰基形成氢键的话,也更有利于反应的进行。这也是日本科学家考虑问题的可怕之处,总是考虑到特别特别细节之处。
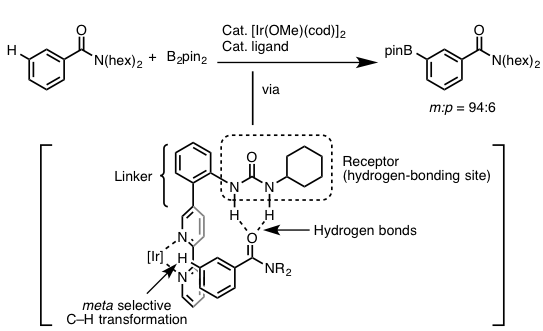
作者利用这一设计的催化剂,也讨论了底物的一般性,对于安息香酸的酯,酰胺等可以与催化剂形成氢键的一系列芳香环,都可以得到间位选择性的硼基化产物,具体的请查阅原论文。
综上就是这回要介绍的通过催化剂控制的芳香环的间位选择性C-H硼基化反应。其实个中辛苦与不断地努力只有当事人自己清楚。对于我们这些从事催化剂设计的研究人员来说,即使自认为设计的催化剂十分完美了,而当实验验证的时候往往会发现恨不如人意,这是十分正常的。所以在设计的基础上,利用一系列的实验讨论,给出事实根据的前提上,再不断地进化,改良催化剂已成为常态。在这里我们可以看到邻,间,对位三个位置的选择性芳香族C-H键的硼基化已经全部给攻破。那么接下来可能就不局限于硼基化,可能会尝试用于其他的C-H活化反应,这很值得期待。
小编所感
小编也是做这种利用分子间氢键等相互作用实现位置或者化学选择性C-H活化反应的研究。在上一篇介绍银催化化学选择性反应的记事中小编也说过了,现在很多很多研究主要还只是利用立体位阻或者导向基团实现选择性,这类反应对底物有很大的限制,主要是由底物控制的选择性反应,不具有很广的一般性,也可以说已经并不是特别具有难度,特别新潮的研究,因此,这也是JACS与Nat.Chem的区别。本论文利用氢键固定住一端,在另一端金属活性位点最接近的反应点进行反应,实现了选择性间位C-H活化,这种可以说就是靶向的反应。这篇文章为什么没有上nature或者science,小编个人认为还是由于这个间位sp2C-H硼基化反应本身来说新颖性跟难度都不是特别大。如果是一类目前还没有实现的反应的话,比如远端位的sp3C-H活化的化,那么档次绝对是稳稳地。不过这篇文章给了很多正在做这类研究的人很大的勇气跟信心。只能点个赞,催化剂设计真的不容易。
本论文背后的故事
最后,我们请到了本论文作者东京大学・ERATO group leader的國信洋一郎老师给我们讲一些本论文背后的故事。

在冈山大学当助教时候就有了这个构思,到现在论文发表出来,整整花了4年时间。我想代表这篇论文的共同作者东京大学药学研究科的金井求教授,博士生井田悠与特任研究员西光海三人,与研究室的所有人分享一下我们的喜悦与激动。
其实本研究的动机早在我大学时代1,2年级的时候就已经萌芽了,那会儿就想着将来做一些C-H键的变换反应研究,然后在冈山大学高井和彦教授的研究室当助教那段时间,体会到了导向基在选择性上的威力,但是另一方面又在想如果一直使用这些导向基的话,在实用性与原创性上大打折扣,心中的疑问也慢慢的多了起来。
在导向基应用上,除了对底物限制性很大这个问题以外,基本上选择性也一般只是出在芳香环的邻位上,间位与对位的选择性反应一般是很难实现的。当然,利用电效应进行的间位选择性反应的例子也有,但是很难控制,有很大的限制因素,所以并不能说已经完全攻克了这类反应。所以我就萌发出了要在不利用导向基的情况下,开发出一种新方法达到间位选择性反应的想法。
对于间位选择性变换的手法,近年来Scripp研究所的余金权教授等人通过不懈的努力开发出的利用长链导向基实现了一系列底物的选择性C-H活化反应被刷刷刷的发表在各大知名期刊上。一开始我也想follow一下那种想法,但是还是没有走出导向基这个限制区域,当然实用性也肯定有所不足,最终还是放弃了那个想法。接着我就有了利用生物酵素通过与底物间形成氢键等非共有结合键的相互作用来实现选择性的想法。在冈山大学的那段时间里,我就开始研究利用非共有结合的相互作用(Lewis酸-碱相互作用),来实现位置选择性的C-H官能团化,这也为我现在这个研究奠定了一定的基础。
实际上,我在2012年4月转到东京大学后,才与从别的大学进来读硕士课程的井田、跟我一起来东京大学做研究员的冈山大学时候的学生西君开始整个实验上的尝试论证。从论文中看那个配体的结构十分简单明了,但是其中我们其实尝试了无数个类似的配体,几度失败后重新设计,重新实验尝试,才最终得到了这么好的结果,这跟这两个学生的不懈努力是分不开的,在这里我衷心的感谢他们俩,尤其是在最低谷时候,身心很疲惫的状态下继续坚持的决心与态度。另外我也要感谢一直以来给了很多很好的建议,并且一直支持这个研究的金井教授。
今后,我们打算把这个concept运用到其他位置的C-H变换反应或者C-H活化以外的反应。C-H键活化反应现在确实是很热,很潮流的研究,但是怎样把它能够发展到烯烃复分解反应、偶联反应那样,真正能够变成很实用,很应用化还有很长的路要走,这也需要所有科研人员的不懈努力,我也会朝着那个目标奋进。
國信 洋一郎
参考文献
- Chen, X; Engle, K. M.; Wang, D.-H.; Yu, J.-Q. Angew. Chem. Int. Ed. 2009, 48, 5094. DOI: 10.1002/anie.200806273
- Colby, D. A.; Bergman, R. G.; Ellman, J. A. Chem. Rev. 2010, 110, 624. DOI: 10.1021/cr900005n
- Cho, J.-Y.; Tse, M. K.; Holmes, D.; Maleczka, R. E. Jr; Smith, M. R. III. Science 2002, 295, 305. DOI: 10.1126/science.1067074
- Phipps, R. J.; Gaunt, M. J. Science 2009, 323, 1593. DOI: 10.1126/science.1169975
- Leow, D.; Li, G.; Mei, T.-S.; Yu, J.-Q. Nature 2012, 486, 518. DOI:10.1038/nature11158
- Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N. R.; Hartwig, J. F. J. Am. Chem. Soc. 2002, 124, 390. DOI:10.1021/ja0173019
- Etter, M. C.; Urbañczyk-Lipkowska, Z.; Zia-Ebrahimi, M.; Panunto, T. W. J. Am. Chem. Soc. 1990,112, 8415. DOI: 10.1021/ja00179a028
- Kawamorita, S.; Ohmiya, H.; Hara, K.; Fukuoka, A.; Sawamura, M. J. Am. Chem. Soc. 2009, 131, 5058. DOI: 10.1021/ja9008419
- Saito, Y.; Segawa, Y. Itami, K J. Am. Chem. Soc. 2015, 137, 5193. DOI: 10.1021/jacs.5b02052
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载