
本文作者:杉杉
导读
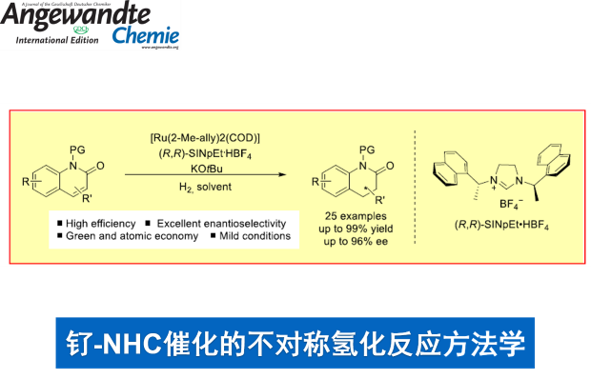
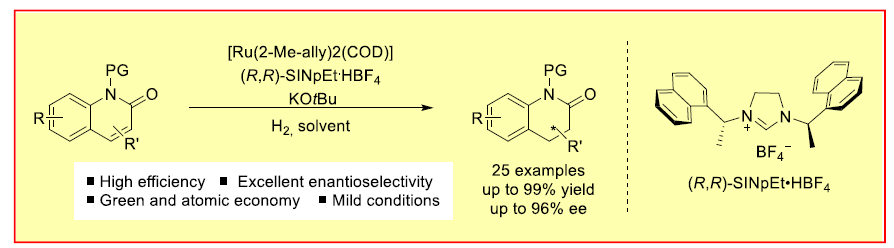
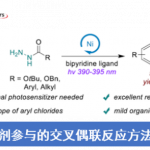
不饱和化合物的直接不对称氢化反应方法学,目前已经成为合成化学中构建手性三维骨架 (three-dimensional motifs)的最为简洁,并且较为关键的策略之一。近日,德国Münster大学F. Glorius课题组在Angew. Chem. Int. Ed.中发表论文,报道一种在温和的反应条件下,采用Ru(II)-NHC催化体系,成功实现一系列2-喹诺酮类化合物 (2-quinolones)参与的不对称氢化反应方法学,进而以较高的反应收率与中等至优良的对映选择性获得一系列相应的烷基、芳基以及卤素取代的光学活性二氢-2-喹诺酮衍生物。同时,这一全新的对映选择性氢化策略为简单手性3,4-二氢-2-喹诺酮类化合物的构建开辟出一条较为有效,并具有良好原子经济性的反应路线。此外,目标产物手性3,4-二氢-2-喹诺酮能够通过进一步的还原过程,最终获得四氢喹啉以及八氢喹诺酮衍生物。
Ru-NHC Catalyzed Asymmetric Hydrogenation of 2-Quinolones to Chiral 3,4-Dihydro-2-Quinolones
T. Hu, L. Lückemeier, C. Daniliuc, F. Glorius, Angew. Chem. Int. Ed. ASAP. doi: 10.1002/anie.202108503.
正文
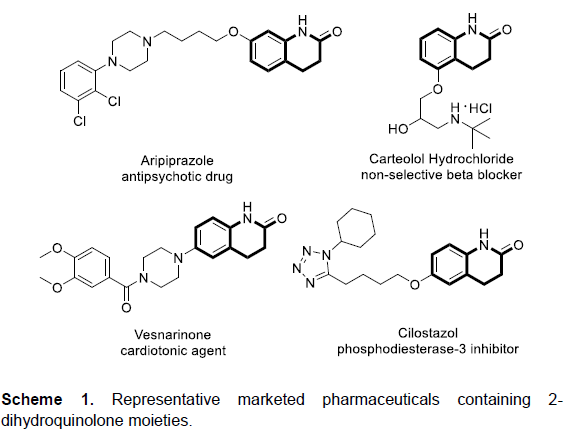
二氢喹诺酮类化合物 (dihydroquinolones)广泛存在于一系列天然产物以及市售的药物中,是一类极为重要的杂环分子,并表现出良好的生物活性。例如,抗精神病药 (antipsychotic drug) aripiprazole、非选择性β受体阻滞剂 (nonselective beta blocker) carteolol、强心剂 (cardiotonic agent) vesnarinone、磷酸二酯酶-3抑制剂(phosphodiesterase-3 inhibitor) cilostazol以及melosuavne等 (Scheme 1)。同时,作为重要的合成中间体,二氢喹诺酮类化合物能够进一步转化为其它几种常见的杂环化合物,例如四氢喹啉以及八氢喹诺酮类化合物。
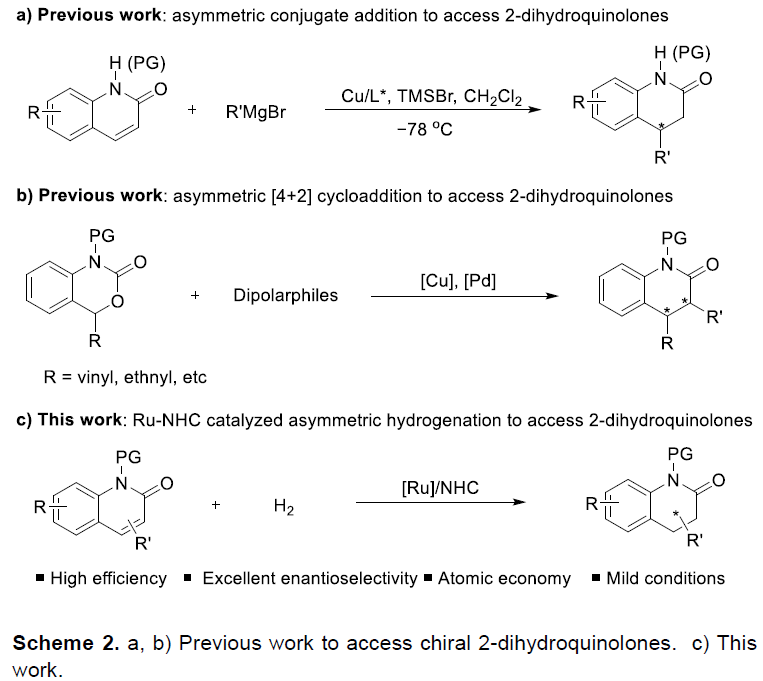
尽管构建相应非手性以及外消旋二氢喹诺酮类化合物的合成设计策略已经有相关的文献报道[1],然而,实现具有光学活性的二氢喹诺酮类化合物,尤其是二氢-2-喹诺酮类化合物的构建,则较少有相关的文献报道。目前,实现手性二氢-2-喹诺酮类分子的构建主要涉及如下两种反应策略:第一种是采用过渡金属催化的不对称共轭加成反应方法学[2]。然而,在这一策略中,多数实例关注于芳基化反应的相关研究。其中,2019年,Harutyunyan课题组[3]通过Grignard试剂参与的不对称烷基化反应方法学,进而成功实现一系列烷基取代的二氢-2-喹诺酮类化合物的合成。然而,由于2-喹诺酮类化合物较低的反应活性,因此,上述策略中,反应条件较为苟刻,同时底物应用范围十分有限 (Scheme 2a)。第二种则是由Cao[4]、Gong[5]以及Xiao[6]课题组独立发展的不对称[4+2]环加成策略,并且,通过这一策略,能够顺利实现一系列二氢-2-喹诺酮类分子的构建。然而,上述策略则需要相关底物中存在特定类型的官能团,例如乙烯基或乙炔基 (Scheme 2b)。因此,开发一种更为通用,并具有高度原子经济的合成转化策略,进而完成各类简单二氢-2-喹诺酮分子的构建,仍然面临诸多的挑战。
而喹诺酮衍生物的直接氢化策略,是构建具有二氢喹诺酮骨架的生物活性分子的最为简洁与最具原子经济性的重要策略之一,同时,在工业大规模合成中,具有良好的应用前景。令人意外的是,2-喹诺酮类化合物,尤其是简单2-喹诺酮类化合物的直接不对称氢化策略,在二氢喹诺酮类化合物合成中的应用,却极少有相关的研究报道[7]。研究表明,由于2-喹诺酮类化合中存在具有较低反应活性的环状α,β-共轭酰胺单元以及酰胺氮原子对于过渡金属中心的毒化效应 (poisoning effect) [8],进而极大阻碍上述氢化过程的有效进行。
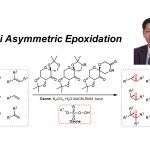
在过去的几十年中,有机合成化学家已经成功设计出多种重要的不对称加氢催化反应体系[9]。其中,本课题组[10]成功发展出一种Ru-NHC配合物催化剂,并将其应用于多种杂环芳香化合物以及非芳环烯基化合物的不对称加氢过程,同时,表现出优良的对映选择性控制。受到这一研究报道的启发,本文中,作者进一步将Ru-NHC配合物催化体系应用于2-喹诺酮类化合物的直接不对称氢化反应方法学的研究 (Scheme 2c)。
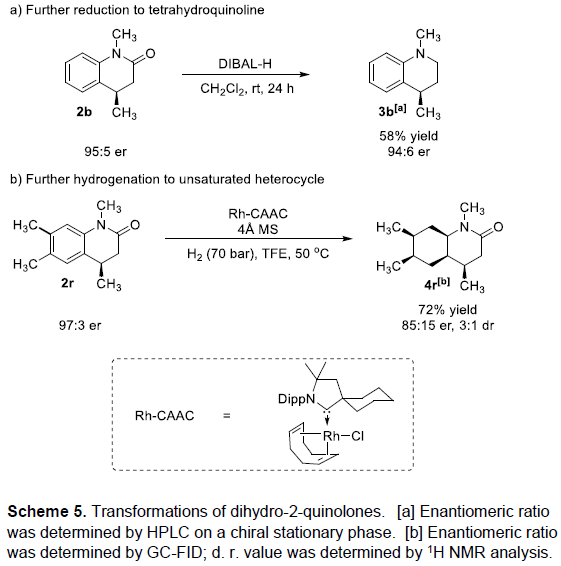
首先,作者采用1a或1b作为模型底物,进行相关反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用Ru(COD)(2-methylally)2作为催化剂,(R,R)-SINpEt•HBF4作为NHC前体,KOtBu作为碱,Et2O作为反应溶剂,氢气压力为10 bar,反应温度为15 oC,最终获得99%收率以及95:5 e.r.的手性产物2b。值得注意的是,由于未保护的喹诺酮底物1a能够通过互变异构化过程,转化为更加稳定的喹诺-2-醇 (quinolin-2-ol),同时,鉴于游离酰胺基团对于催化剂中心毒化作用的存在,这里,作者选择N-甲基保护的2-喹诺酮2b作为反应底物。
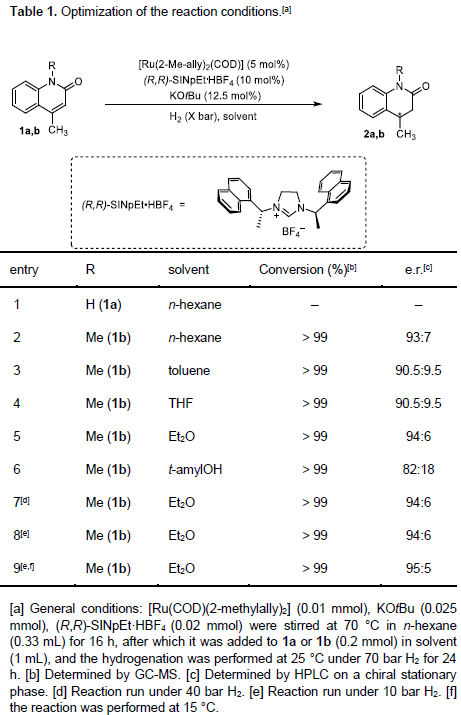
在上述的最佳反应条件下,作者首先对各类2-喹诺酮底物的应用范围进行考察 (Scheme 3)。研究表明,N-苄基保护的2-喹诺酮底物,同样能够以98%的分离收率以及95:5 e.r.,获得预期的手性产物2c。同时,作者发现,上述的标准反应条件对于在6-位中具有烷基、卤素基团、甲氧基以及苯基取代的2-喹诺酮底物,同样能够以较高的收率以及良好至优良的对映选择性,获得相应的手性产物2d–2k。值得注意的是,在上述的最佳反应条件下,未观察到去卤化产物的形成。同时,对于6-位具有甲氧基取代的2-喹诺酮底物,由于双键中取代基电子性质的影响,进而使产物2i的收率出现显著降低。
之后,该小组发现,上述的最佳反应条件对于7-位中具有烷基、苯基、卤素基团以及甲氧基取代的2-喹诺酮底物,同样能够较好地兼容,并以较高的反应收率与优良的对映选择性,获得相应目标产物2l–2q以及2t–2u。接下来,作者进一步观察到,上述的标准反应体系对于具有双重甲基取代的2-喹诺酮底物,同样能够有效地兼容,并获得相应手性产物2r与2s。此外,该小组发现,通过这一全新的对映选择性氢化策略,能够以99%的收率与高度的对映选择性,获得相应的生物活性分子2v。同时,作者进一步通过X-射线晶体学分析,最终确定产物2v的绝对构型为R–构型。
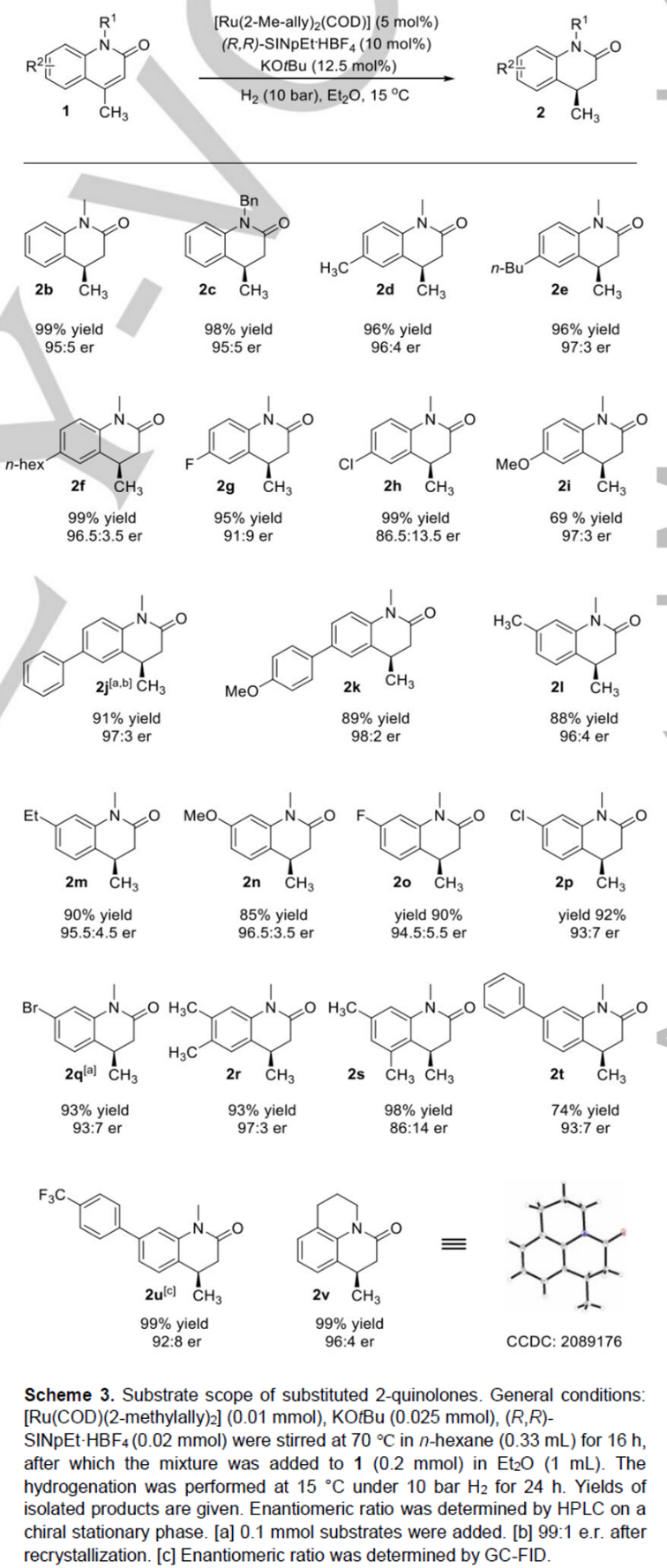
接下来,作者对4-位取代的2-喹诺酮底物的应用范围进行深入研究 (Scheme 4)。该小组发现,4-乙基取代的2-喹诺酮底物1w,同样能够有效地参与上述的不对称氢化过程,进而获得相应手性产物2w,收率为99%,e.r.为77:23。同时,研究发现,上述的标准反应条件对于4-位具有更高立体位阻基团取代的2-喹诺酮底物,例如1x与1y,同样能够以优良的反应收率与良好的对映选择性,获得相应的手性产物2x与2y。并且,较为有趣的是,3-甲基取代的喹诺酮底物2z,同样能够与上述的标准条件良好地兼容,并获得较好的反应收率与优良的对映选择性。
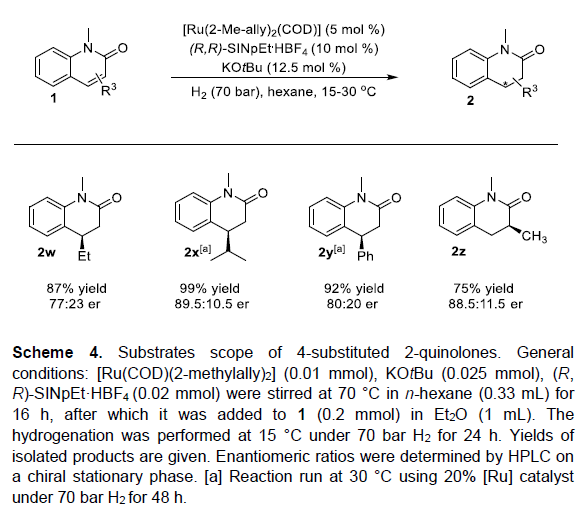
为进一步阐明这一全新的对映选择性氢化策略的合成实用性,作者对获得的手性产物进行相关的后期修饰研究 (Scheme 5)。该小组观察到,二氢-2-喹诺酮产物2b在DIBAL-H还原剂存在的条件下,能够进一步转化为相应的四氢喹啉化合物3b,并且,反应过程中无相应对映体光学纯度的丧失。之后,作者发现,具有二甲基取代的手性产物2r在Rh-CAAC/H2催化体系中,最终能够获得72%收率的八氢喹诺酮化合物4r,e.r.为85:15,dr为3:1。
总结:德国Münster大学的F. Glorius课题组报道首例采用钌-NHC催化剂促进的简单2-喹诺酮类化合物的不对称氢化反应方法学,进而以极高的反应收率 (高达99%)与中等至优良的对映选择性 (高达98:2 er),获得相应的3,4-二氢-2-喹诺酮衍生物。这一全新的对映选择性氢化策略具有良好的官能团兼容性。同时,反应过程能够在较为温和的条件下进行。此外,相应的手性3,4-二氢-2-喹诺酮产物同样能够经历进一步的合成转化过程,进而获得一系列较为重要的手性三维骨架。
参考文献:
[1] a) M. Castaing, S. L. Wason, B. Estepa, J. F. Hopper, M. C. Wills, Angew. Chem. Int. Ed. 2013, 52, 13280. doi: 10.1002/anie.201308127.b) J. Yan, H. Li, X.W. Liu, J. L. Shi, X. Wang, Z. J. Shi, Angew. Chem. Int. Ed. 2014, 53, 4945. doi: 10.1002/anie.201402562.
c) K. Wang, X. Chen, M. Yuan, M. Yao, H. Zhu, Xue, Z. Luo, Y. Zhang, J. Org. Chem. 2018, 83, 1525. doi:10.1021/acs.joc.7b02585.
[2] a) J. F. Paquin, C. R. J. Stephenson, C. Defieber, E. M. Carreira, Org. Lett. 2005, 7, 3821. doi: 10.1021/ol051533l.b) L. Zhang, Z. Qureshi, L. Sonaglia, M.Lautens, Chem. Int. Ed.2014, 53, 13850. doi: 10.1002/anie.201407400.
[3] Y. Guo, S. R. Harutyunyan, Angew. Chem. Int. Ed. 2019, 58, 12950. doi: 10.1002/anie.201906237. [4] X. Lu, L. Ge, C. Cheng, J. Chen, W. Cao, X. Wu, Chem. Eur. J. 2017, 23, 7689. doi: 10.1002/chem.201701741. [5] J. Song, Z. J. Zhang, L. Z. Gong, Angew. Chem. Int. Ed. 2017, 56, 5212. doi: 10.1002/anie.201700105. [6] Y. Zhao, J. R. Chen, W. J. Xiao, Org. Lett. 2016, 18, 6304. doi: 10.1021/acs.orglett.6b03174.b)M. Li, Y. Wei, J. Liu, H. W. Chen, L. Q. Lu, W. J. Xiao, J. Am. Chem. Soc. 2017, 139, 14707. doi: 10.1021/jacs.7b08310.
c) Y. Wang, Q. Xiong, L.Q. Lu, Q. L. Zhang, Y. Wang, Y. Lan, W. J. Xiao, Angew. Chem. Int. Ed. 2019, 58, 11013. doi: 10.1002/anie.201905993.
[7] Q. Zhao, X. Wu, F. Yang, P. Yan, J. Xie, Q. Zhou, Org. Lett. 2021, 23, 3593. doi: 10.1021/acs.orglett.1c00993. [8] W. Li, C. Schlepphorst, C. Daniliuc, F. Glorius, Angew. Chem. Int. Ed. 2016, 55, 3300. doi: 10.1002/anie.201512032. [9] a) D. S. Wang, Q. A. Chen, S. M. Lu, Y. G. Zhou, Chem. Rev. 2012, 112, 2557. doi: 10.1021/cr200328h.b) J. J. Verendel, O. Pamies, M. Dieguez, P. G. Anderson, Rev. 2014, 114, 2130. doi:10.1021/cr400037u.
c) Z. Zhang, N. A. Butt, W. Zhang, Rev. 2016, 116, 14769. doi:10.1021/acs.chemrev.6b00564.
[10] a) S. Urban, N. Ortega, F. Glorius, Angew. Chem. Int. Ed. 2011, 50, 3803. doi: 10.1002/anie.201100008.b) S. Urban, B. Beiring, N.Ortega, D. Paul, F. Glorius, Am. Chem. Soc. 2012, 134, 15241. doi:10.1021/ja306622y.
c) N.Ortega, S. Urban, B. Beiring, F. Glorius, Chem. Int. Ed. 2012,51, 1710. doi: 10.1002/anie.201107811.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!




















No comments yet.