
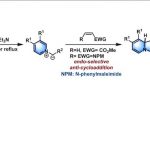
上一期小编介绍了由日本Kyushu大学的Matsuda (松田 昴,九州大学工学部合成化学科, Matsuda Tsutomu, Department of Organic Synthesis, Faculty of Engineering,
Kyushu University)研究室发展的Heck-Matsuda偶联反应方法学[1]。该反应为各类烯基化合物的芳基化开辟了一种全新的方法。然而,反应过程中需要使用化学计量且具有潜在爆炸性的芳香重氮盐,限制了该反应在工业生产中的应用。同时,该反应对空气与湿气较为敏感,同样使该方法学的应用受到限制。为进一步提高该反应的应用范围,诸多课题组对Heck-Matsuda偶联方法学的催化剂、溶剂等条件进行了进一步改进。这里小编将对此进行详细介绍。
概要
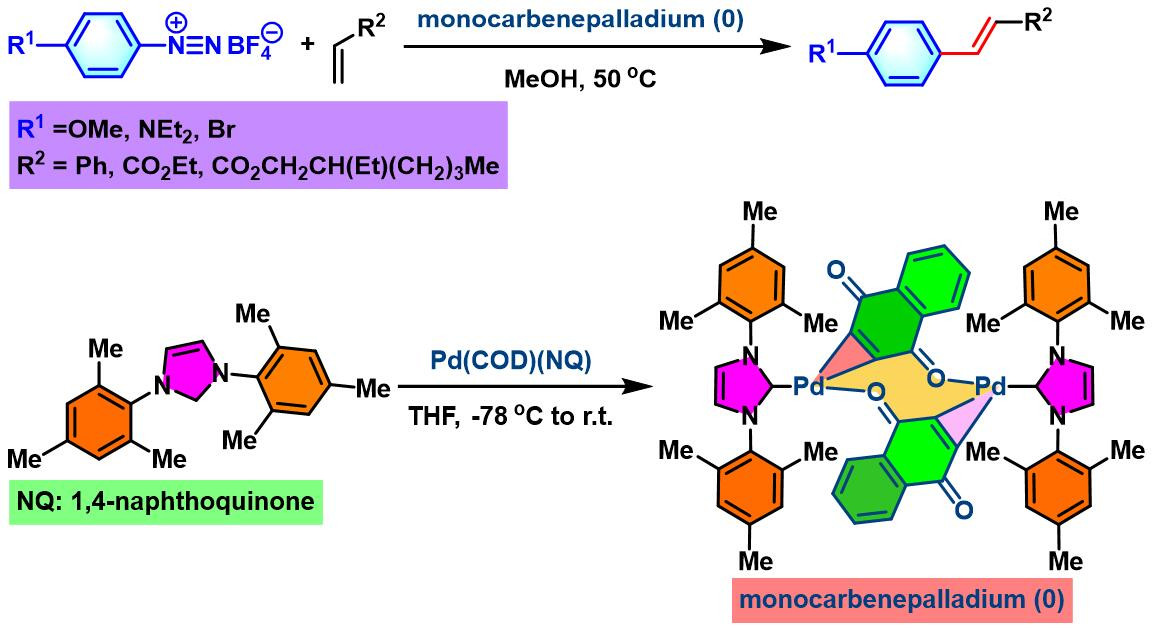
2002年,M. Beller通过[(cod)Pd(NQ)]与carbene配体之间的配体取代过程,合成出新的monocarbenepalladium (0)催化剂,并将其应用于芳香重氮盐与烯烃之间的交叉偶联,研究表明该催化剂能够极大提高反应收率[2]。
2003年,Hu发现通过Li2PdCl4/CuCl催化体系,可以完成羧基取代的芳香重氮盐与丙烯酸酯的Heck-Matsuda偶联 [3]。
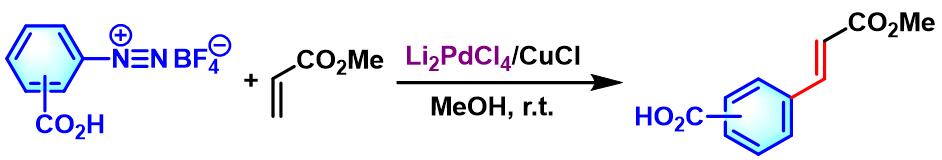
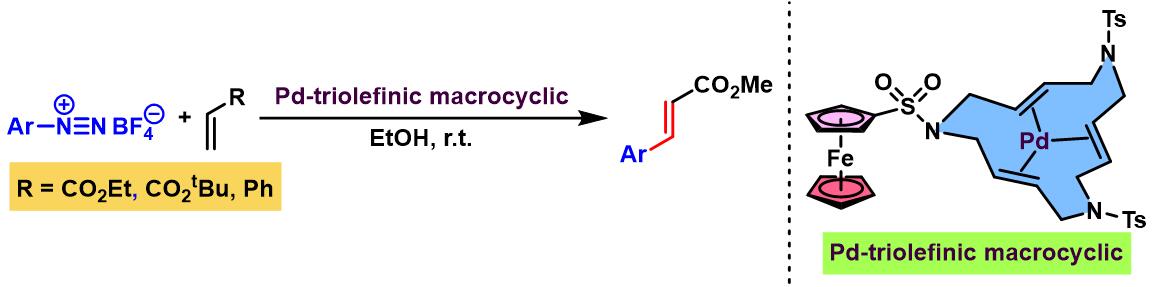
同时,A. Roglans采用更稳定的钯-大环三烯 (Pd-triolefinic macrocyclic)催化剂,使偶联过程能够在更加温和的条件下进行,同时,A. Roglans发现该催化剂还可以在氧气与湿气存在的情况下使用[4]。
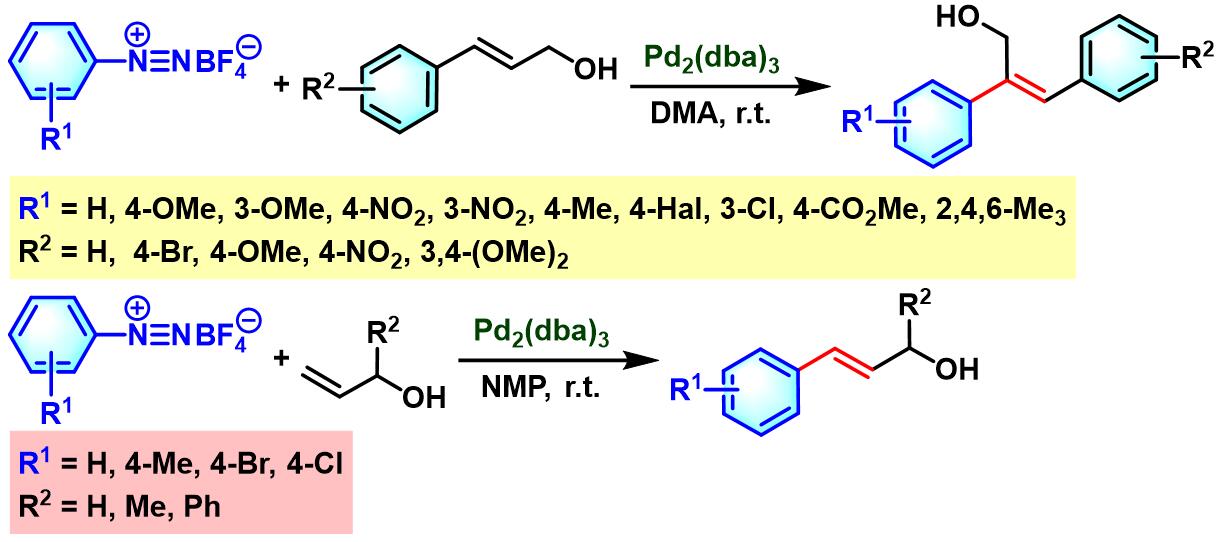
2005年,J. Muzart采用Pd(dba)2催化剂,在无其它配体及碱存在的情况下,系统地研究了芳香重氮盐与各类烯丙醇之间的偶联反应。J. Muzart发现,二级烯丙醇参与的偶联过程,最终获得β-芳基取代的羰基化合物;而一级烯丙醇参与的偶联反应,却获得芳基化缩醛产物[5]。

2006年,M. Barbero将新发展的邻苯二磺酰亚胺芳香重氮盐应用于钯催化的一级与二级烯丙醇的芳基化[6]。研究表明,一级与二级烯丙醇在该条件下的芳基化过程,均获得β-芳基取代的羰基化合物。
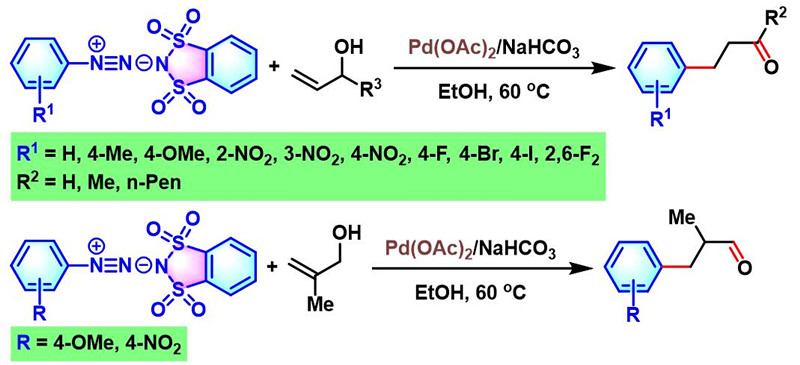
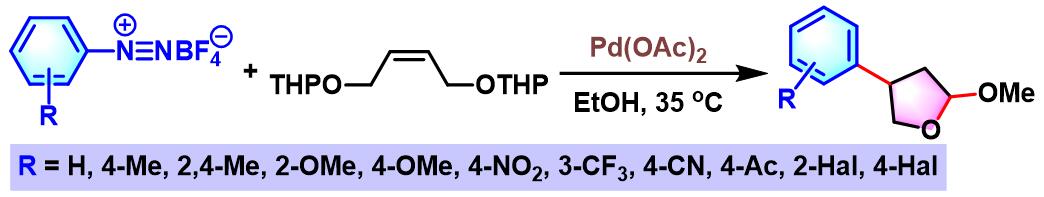
2009年,S. Cacchi将烯基底物进一步扩展至THP保护的(Z)-2-丁烯-1,4-二醇,并顺利完成各类β-芳基-γ-丁内酯化合物的构建[7]。
之后,S. Cacchi进一步通过4-羟基-2-丁烯酸甲酯与芳香重氮盐之间的偶联反应,完成4-芳基丁烯酸内酯的合成[8]。
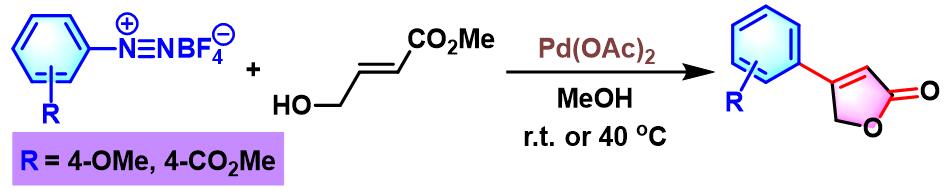
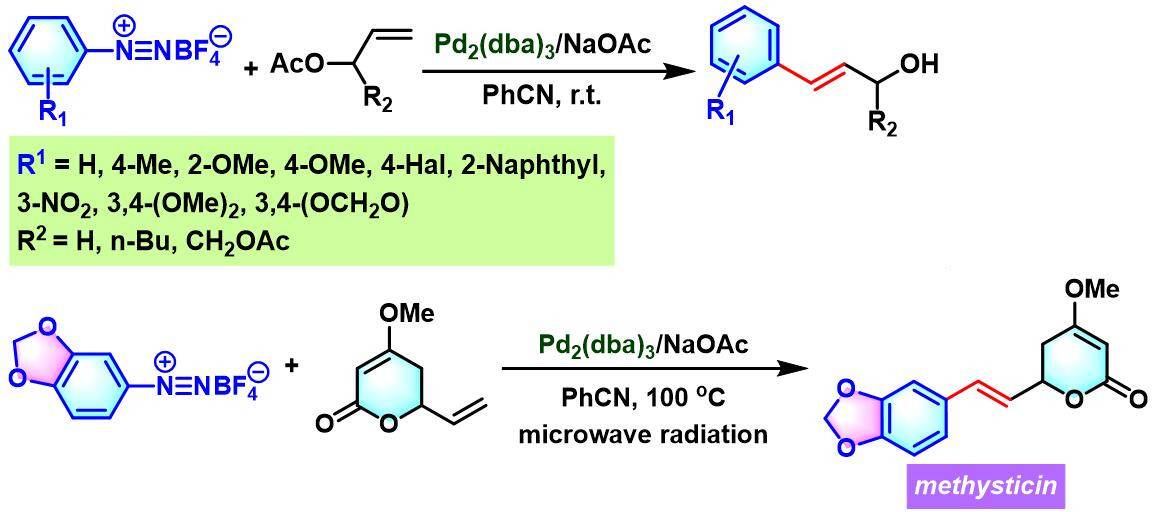
C. R. D. Correia课题组继续对烯丙酯参与的Heck-Matsuda反应进行了细致研究,并将其应用于methysticin的合成[9]。
2010年,B. Schmidt小组将芳香重氮盐的应用范围扩展至具有酚羟基取代芳香重氮盐[10]。
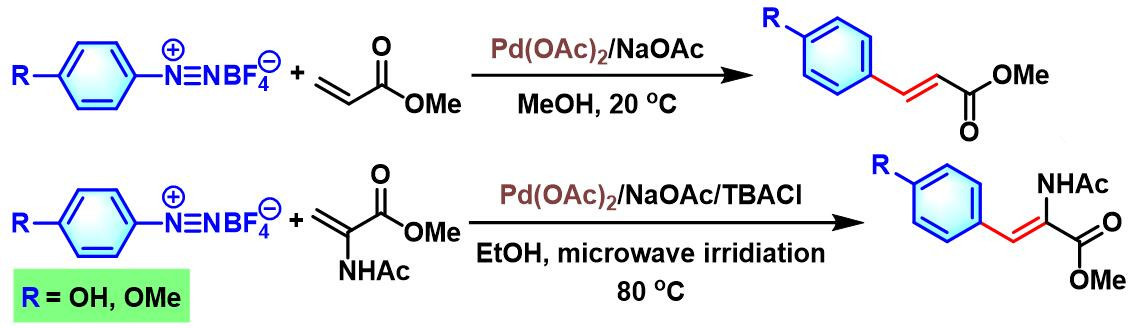
2011年,A. Vallribera进一步将Heck-Matsuda偶联方法学扩展至对位全氟辛基取代的三氟乙酸芳香重氮盐,并顺利地构建起一系列含氟芳香化合物[11]。
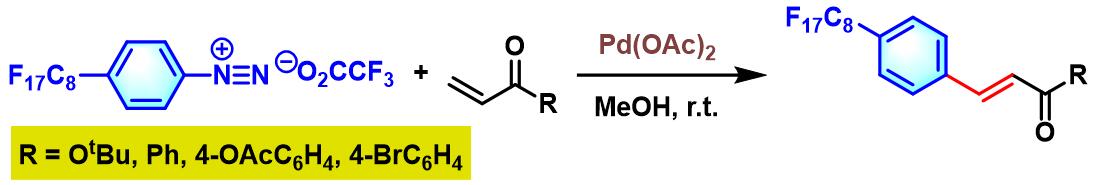
同年,C. R. D. Correia课题组发现,2-杂原子取代的丙烯酸酯与芳香重氮盐在钯催化的条件下,同样能够使双键的芳基化过程顺利进行[12]。

接下来,C. R. D. Correia发现在乙酸钯催化下,采用供电子基团取代的芳香重氮盐,可以使丙烯酸酯发生双重芳基化[13]。并且,该方法为生物活性化合物indanones的合成设计提供了一种更为简便的途径。

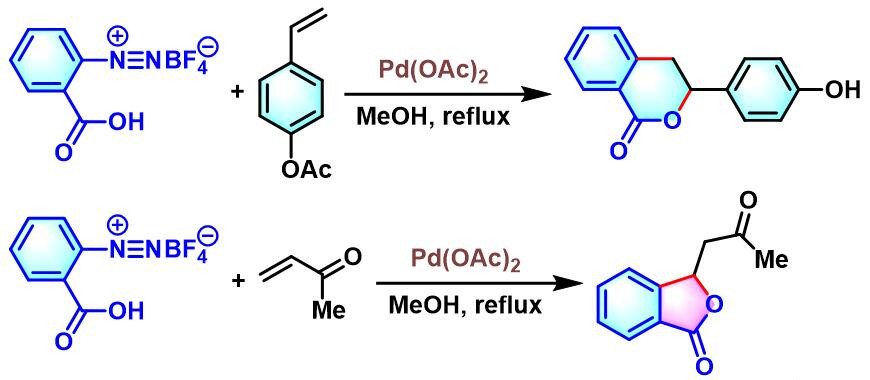
同时,C. R. D. Correia进一步将钯催化条件下,邻位羧基取代的芳香重氮盐与苯乙烯之间的偶联反应,应用于3,4-dihydroisocoumarins及phthalides分子的构建[14]。
而K. K. Laali小组发现,通过[BMIM][PF6]离子液体作为溶剂,可以进一步拓宽烯基底物的应用范围。反应无需加入碱添加剂,离子液体在反应完成后,能够循环利用,并可以获得优良的反应产率与立体选择性[15]。同时,环境友好,适合于工业大规模生产。

F.-X. Felpin课题组根据芳胺与亚硝酸酯能够原位产生芳香重氮盐的反应原理,设计出钯催化剂-甲磺酸双重催化 (bicatalytic approach)循环的策略。该策略避免了直接使用化学计量的芳香重氮盐,进而将反应过程中芳香重氮盐的用量将至亚化学计量 (substoichiometric amount)[16]。因而,提高了实验操作的安全性。之后,D. Jacquemin与F.-X. Felpin共同合作,对这一策略的应用范围进行了进一步阐述[17]。
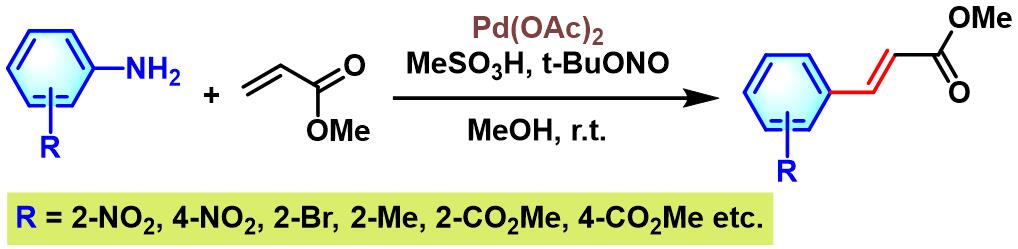
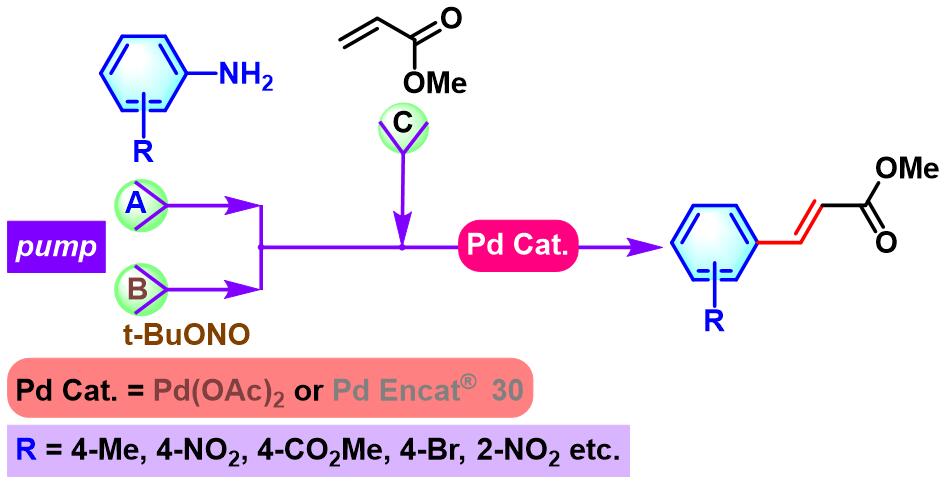
同时,F.-X. Felpin小组采用连续流动反应器 (continuous-flow reactor ),进一步提高了实验操作的简便性及安全性。同时,F.-X. Felpin发现这种连续流动反应器对于均相 (乙酸钯催化剂)与非均相 (Pd Encat® 30催化剂)条件均可适用[18]。之后,该小组通过改进的Nelder-Mead算法 (modified Nelder-Mead algorithm),对连续流动反应条件下的Heck-Matsuda偶联反应进行了进一步优化[19]。
2012年,D. M. Pore研究发现Pd(OAc)2与非离子表面活性剂 (nonionic surfactants) Triton X-100原位形成的Pd NP (Pd nanoparticle)催化剂,同样能够有效地促进芳香重氮盐与烯基化合物之间的交叉偶联反应[20]。而且,在Pd NP催化条件下,具有较高的产率与立体选择性,并对湿气环境不敏感。同时,无需额外添加其他配体,因此,更加的经济友好 (eco-friendly)。

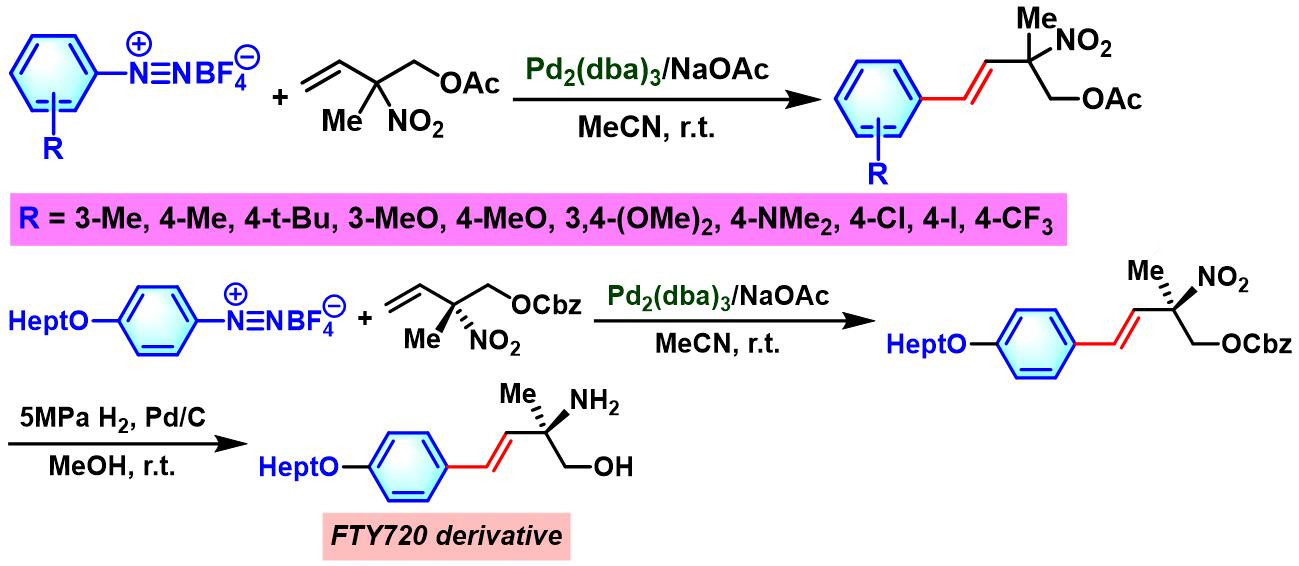
同时,Itoh (伊藤 敏幸, 鳥取大学大学院工学研究科化学•生物応用工学専攻, Itoh Toshiyuki, Department of Chemistry and Biotechnology, Graduate School of Engineering, Tottori University)与Kamimura (上村 明男, 山口大学工学部応用化学科, Kamimura Akio, Department of Applied Molecular Bioscience, Graduate School of Medicine, Yamaguchi University)共同对烯丙基硝基化合物与芳基重氮盐在钯催化条件下的偶联反应进行了详尽地研究,并将其应用于FTY720的不对称合成[21]。
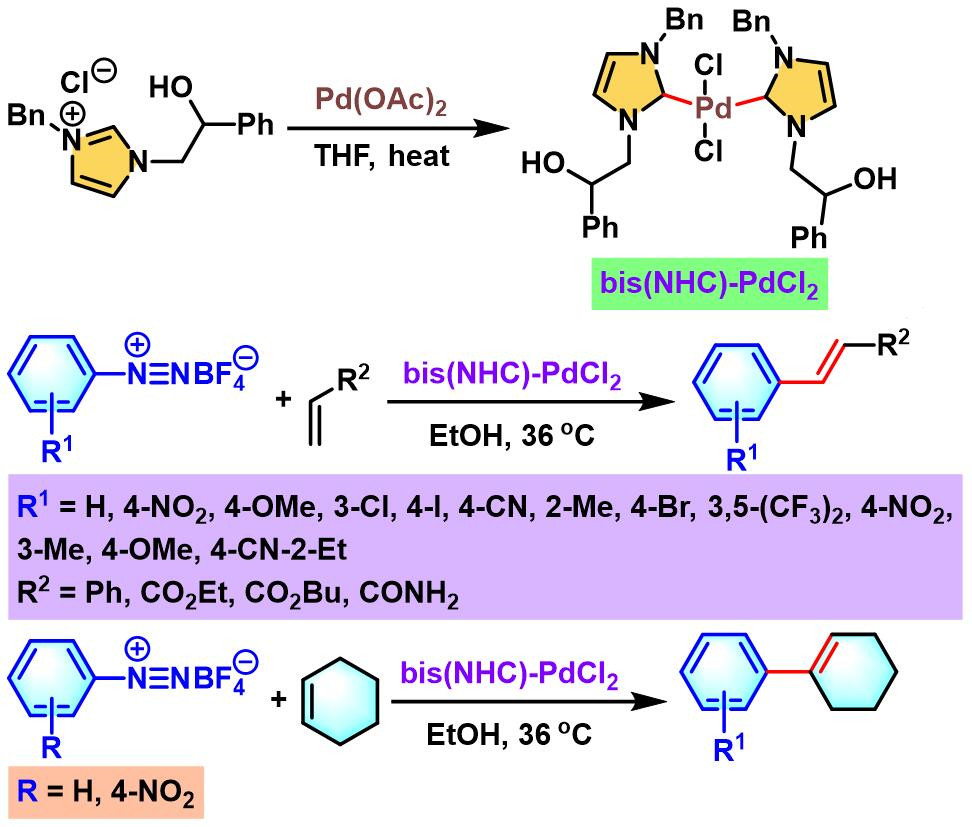
I. M. Pastor进而研究发现,采用羟烷基官能团化的双NHC-Pd催化剂,同样可以完成烯基化合物Heck-Matsuda芳基化[22]。同时,I. M. Pastor发现该催化剂具有更高的转化效率,可以有效降低钯催化剂的用量 (0.5-1 mol%)。
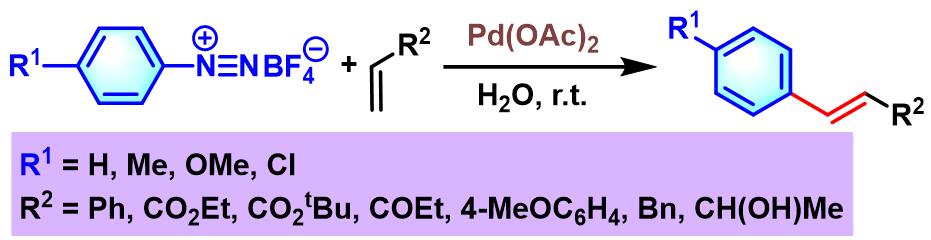
2013年,R. M. Sebastián与A. Vallribera发现在乙酸钯催化剂及水相条件下,芳香重氮盐与烯基化合物之间的偶联反应,仍然能够有效地进行[23]。
之后,M. Gholinejad发现采用agarose负载的Pd NP,同样可以使芳基重氮盐与烯基化合物之间的偶联反应在水相中有效地进行。同时,研究表明,通过agarose负载的Pd NP,还可以有效降低Pd催化剂的用量[24]。
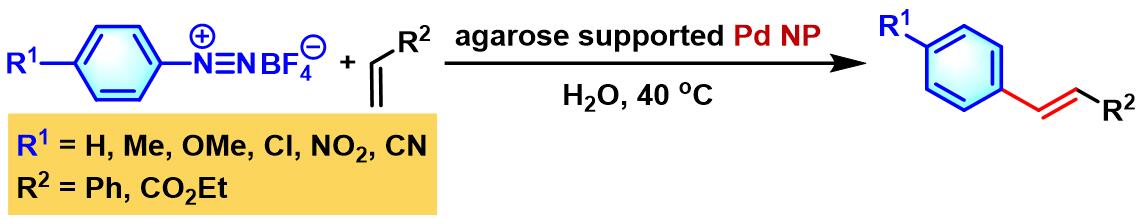
2015年,Li课题组设计出均相的CMC负载Pd NP (Carboxymethylcellulose-supported Pd NP)催化剂。研究表明,选用该催化剂,除可以使Heck-Matsuda偶联反应在水相或水-醇混合溶剂中有效地进行外,还能够使反应条件更加温和且环境友好,并获得良好的收率。而且,该催化剂还可以有效地循环利用[25]。
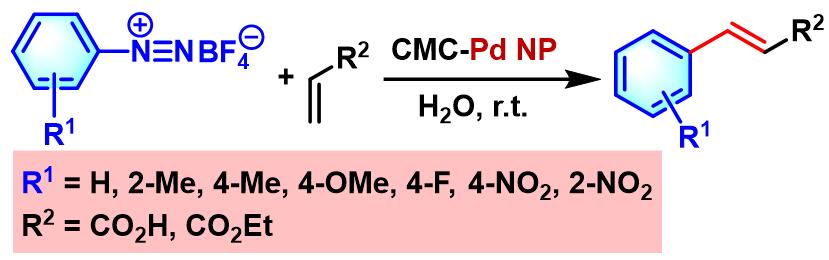
之后,R. Jana通过对烯丙醇底物的立体与电子因素进行系统地研究,成功设计出具有高度区域及立体选择性的烯丙醇与芳香重氮盐之间的Heck-Matsuda交叉偶联反应[26]。
同时,P. S. Postnikov采用新的对甲苯磺酸芳香重氮盐 (ADT, arenediazonium tosylate) 对烯基化合物进行芳基化。实验观察到反应不仅可以在水相中进行,而且,可以获得极高的反应收率。对于部分底物,产率可以接近定量[27]。

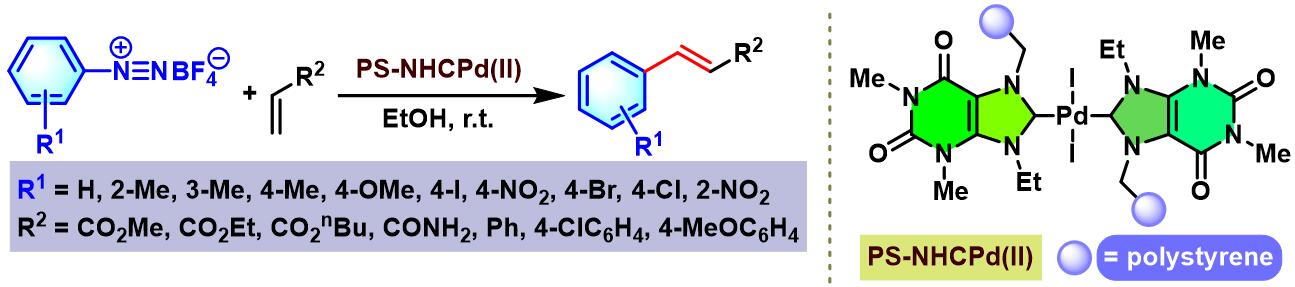
2016年,B. Movassagh研究室采用theophylline作为环境友好型的NHC前体 (NHC precursor),合成出聚苯乙烯负载的Pd-NHC (PS-NHCPd(II), polystyrene-supported palladium(II)-N-heterocyclic carbene complex)异相催化剂[28]。B. Movassagh研究表明,该催化剂具有优良的催化活性与稳定性。同时,能够有效地实现芳香重氮盐与各类烯基化合物之间的交叉偶联反应。
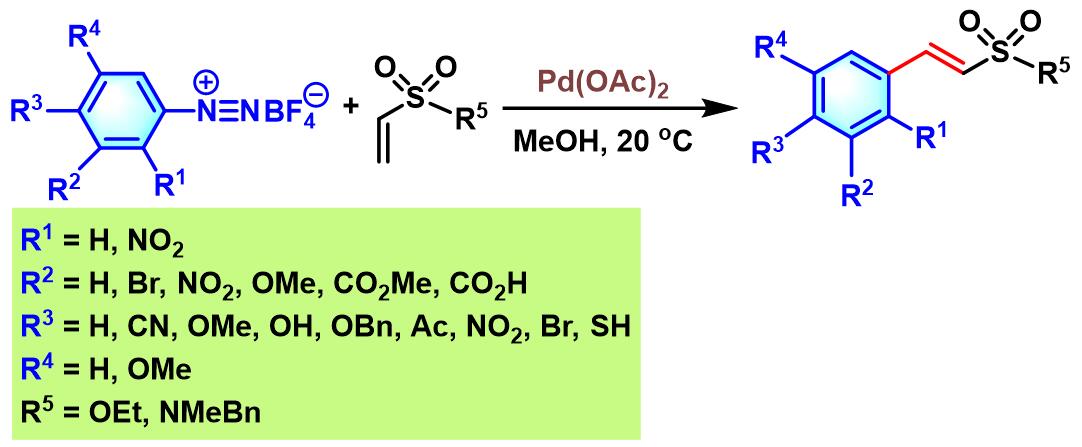
接下来,B. Schmidt将钯催化的芳香重氮盐Heck-Matsuda偶联反应进一步应用于烯基磺酸酯与烯基磺酰胺[29]。
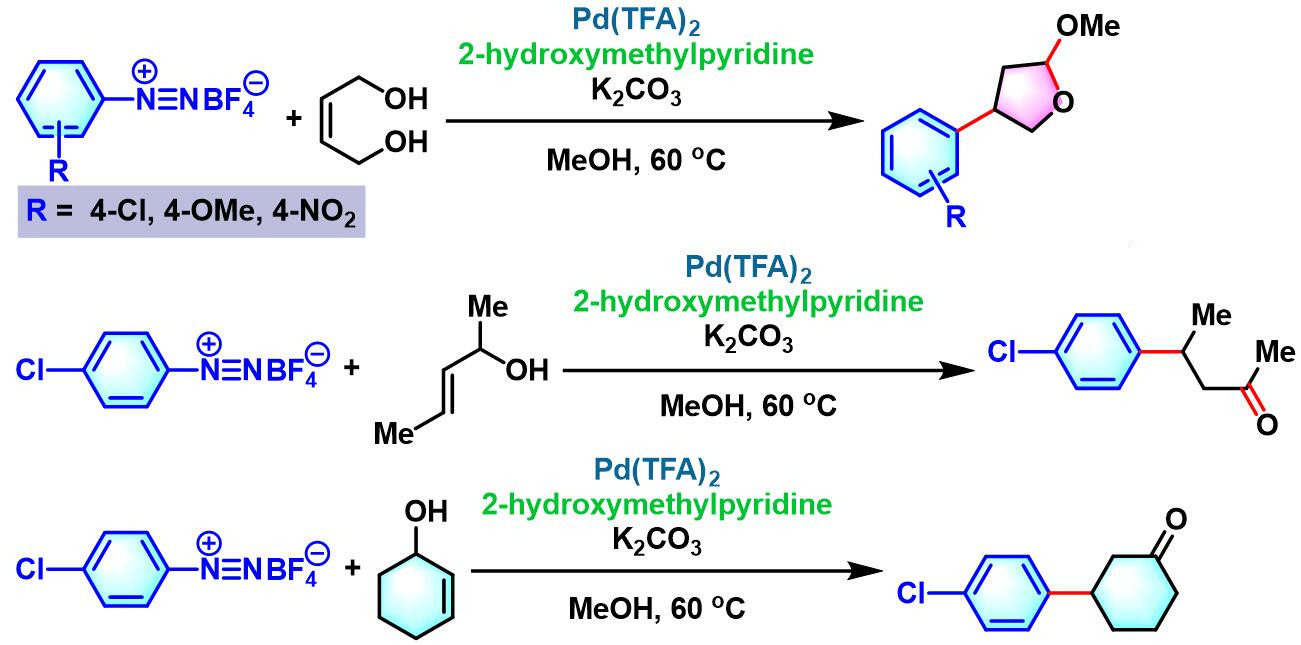
F.-X. Felpin与M. Rodriguez-Zubiri发现通过廉价的2-羟甲基吡啶配体,可以成功地解决烯丙醇底物与芳香重氮盐进行偶联反应时,产率较低的问题[30]。
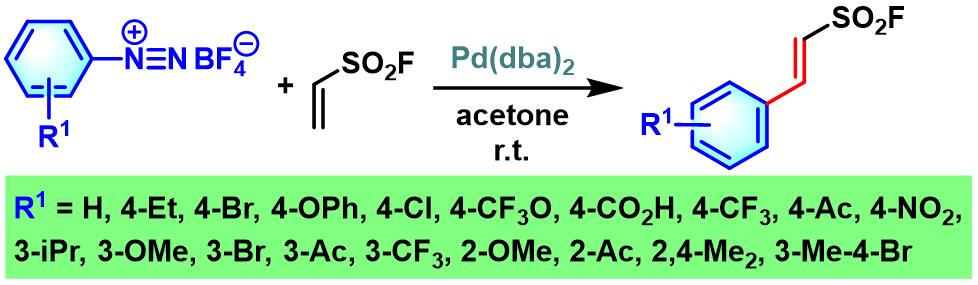
K. B. Sharpless通过芳香重氮盐与乙烯基磺酰氟(ethenesulfonyl fluoride, ESF)之间的Heck-Matsuda偶联反应,顺利完成β-芳乙烯基磺酰氟 (β-arylethenesulfonyl fluoride)的合成[31]。
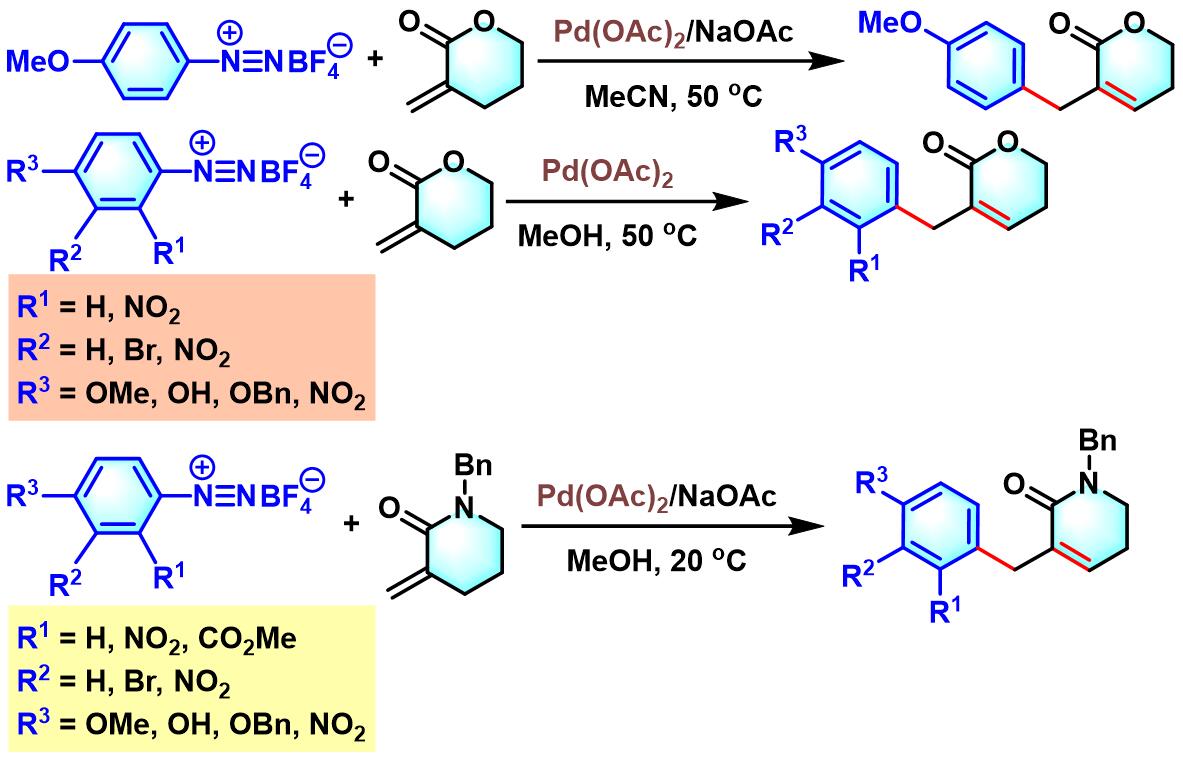
B. Schmidt对钯催化下,α-亚甲基-γ-丁内酯、α-亚甲基-δ-戊内酰胺及α-亚甲基-δ-戊内酯与芳香重氮盐之间的Heck-Matsuda反应进行了系统地研究[32]。B. Schmidt观察到反应区域选择性受到环大小的强烈影响: 六元环的α-亚甲基-δ-戊内酰胺与α-亚甲基-δ-戊内酯可以区域专一性地分别获得α-苄基戊烯酸内酰胺与α-苄基戊烯酸内酯(α-benzyl pentenolide);而五元环的α-亚甲基-γ-丁内酯,可以区域选择性地获得(E)-α-亚苄基-γ-丁内酯 ((E)-α-benzylidene-γ-butyrolactone)产物。同时,作者通过DFT计算研究证实,两种底物采用反应时,区域选择性的差异源于动力学因素。
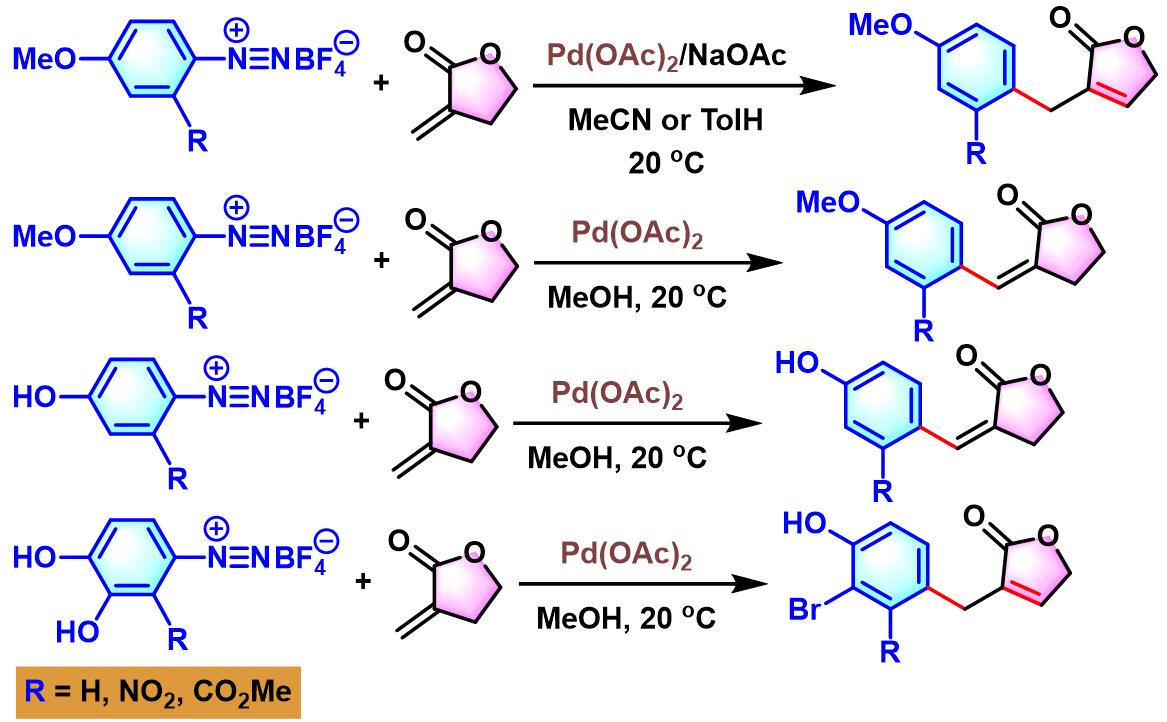
H. Brunner通过乙酸钯与甲醇原位制备的Pd/Al2O3催化剂,成功实现了克级规模的芳香重氮盐与烯基化合物之间的单芳基化与三重芳基化反应[33]。

D. S. Gaikwad通过新开发的prolinate functionalized IL (ionic liqui)-Pd NP催化剂,顺利实现各类芳香重氮盐与烯基化合物之间的Heck-Matsuda反应。D. S. Gaikwad研究发现该催化剂参与的偶联过程具有高度的(E)-立体专一性[34]。
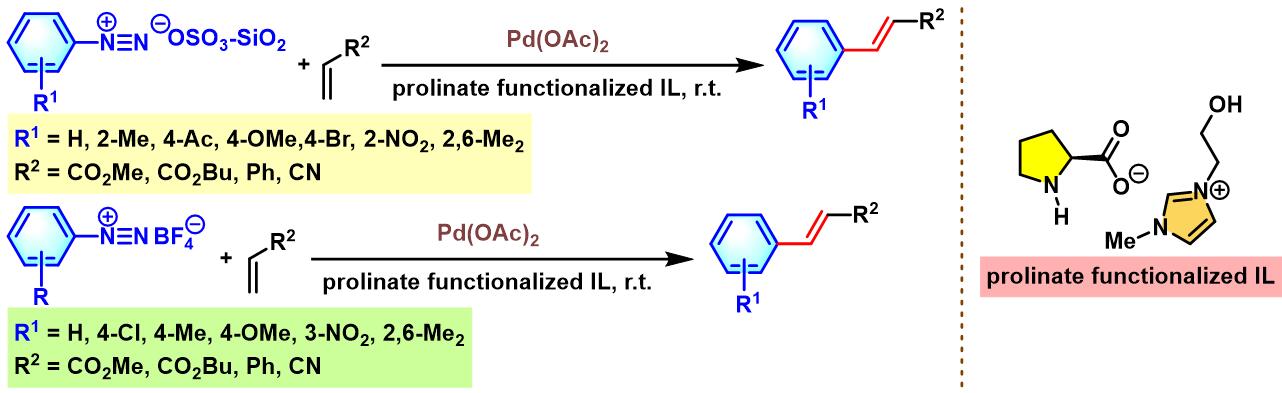
H. Brunner采用原位生成的Pd-磷酸铝催化剂,在更加温和的条件下完成β-二芳基烯基化合物的合成。同时,使反应收率获得较大提高[35]。
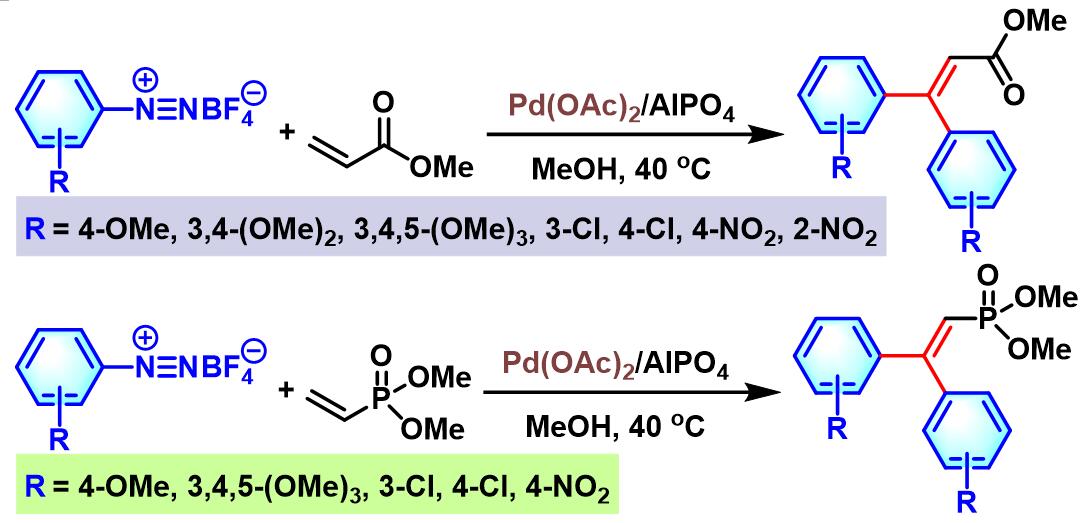
2019年,H. Brunner发现通过层状硅酸钙 (calcium-phyllosilicate) Circosil® (又称为tobermorite, 5 CaO • 6 SiO2 • 5 H2O)或Circolit® (又称为xonotlite, 6 CaO • 6 SiO2 • H2O)同时作为碱与催化剂载体,同样能够顺利实现芳香重氮盐与烯基化合物之间的交叉偶联反应[36]。而且,对于酸敏感产物,如肉桂醛与benzalacetone,在该条件下,同样可以获得优良的产率。
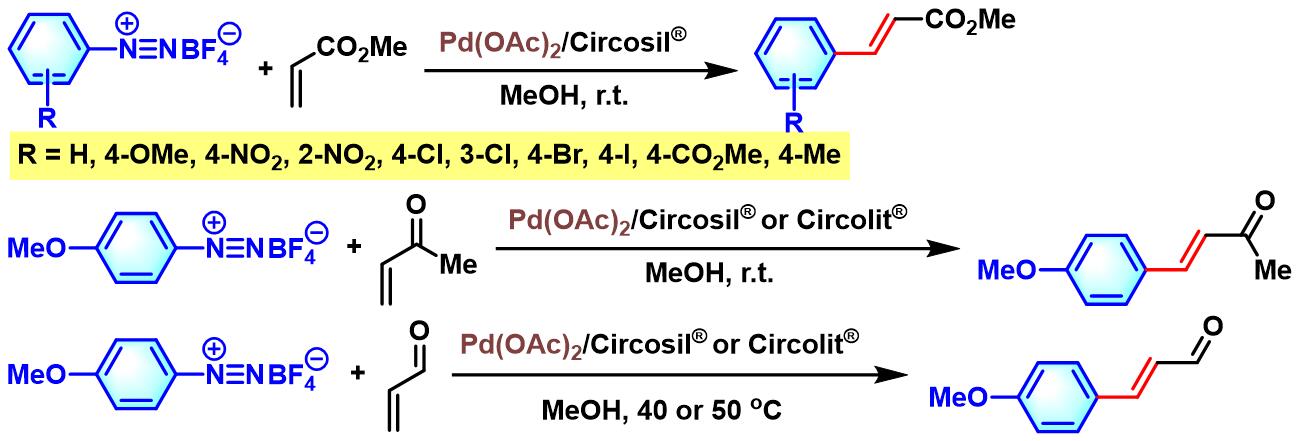
同时,B. Schmidt将钯催化的芳香重氮盐的Heck-Matsuda交叉偶联方法学应用于itaconimide的iterative芳基化 (iterative arylation) [37]。

2020年,H. Brunner采用原位制备的Pd/Al2O3催化剂,顺利完成亚甲基丁二酸酯的芳基化[38]。并且,H. Brunner进一步发现,升高反应温度,可以进一步实现亚甲基丁二酸酯的二芳基化。
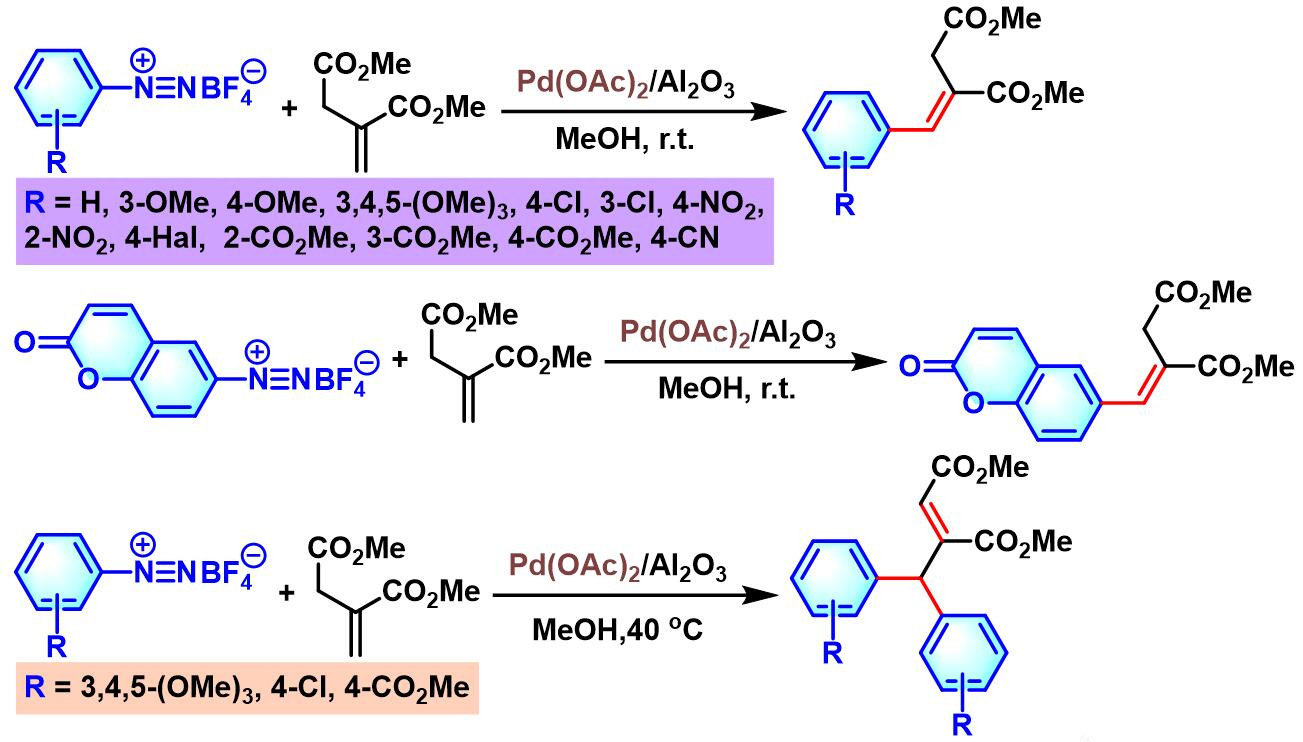
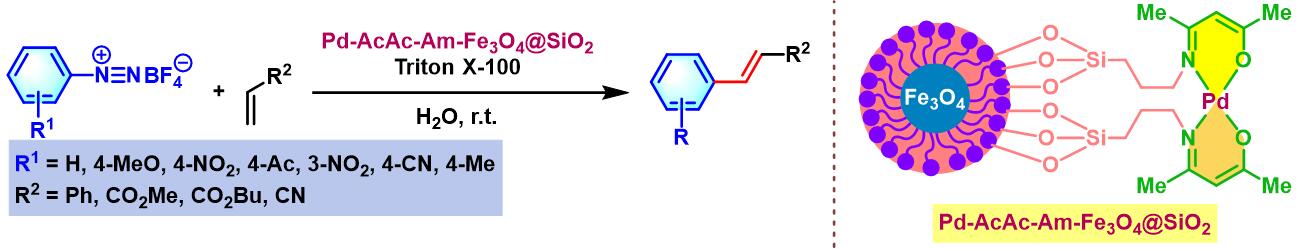
同时,D. M. Pore发现通过固定于胺功能化磁性纳米粒子 (amine functionalized magnetic nanoparticle)中的Pd-Schiff碱催化剂,能够在水相及更加温和的条件下,完成苯乙烯、丙烯酸酯与丙烯腈的Heck-Matsuda芳基化[39]。
基本文献
- [1] K. Kikukawa, T. Matsuda, Chem. Lett. 1977, 159. doi: 10.1246/cl.1977.159.
- [2] K. Selvakumar, A. Zapf, A. Spannenberg, M. Beller, Chem. Eur. J. 2002, 8, 3901. doi: 10.1002/1521-3765(20020902)8:17<3901::aid-chem3901>3.0.co;2-e.
- [3] C. Wang, L. Tan, J. He, H. Hu, J. Xu, Synth. Commun.2003, 33, 773. doi: 10.1081/SCC-120016321.
- [4] J. Masllorens, M. Moreno-Mañas, A. Pla-Quintana, A. Roglans, Org. Lett. 2003, 5, 1559. doi: 10.1021/ol034340b.
- [5] J. Masllorens, S. Bouquillon, A. Roglans, F. Henin, J. Muzart, J. Organomet. Chem. 2005, 690, 3822. doi: 10.1016/j.jorganchem.2005.05.020.
- [6] M. Barbero, S. Cadamuro, S. Dughera, Synthesis 2006, 3443. doi: 10.1055/s-2006-950245.
- [7] S. Cacchi, G. Fabrizi, A. Goggiamani, A. Sferrazza, Synlett 2009, 973. doi: 10.1055/s-0028-1087959.
- [8] S. Cacchi, G. Fabrizi, A. Goggiamani, A. Sferrazza, Synlett 2009, 1277. doi: 10.1055/s-0028-1088132.
- [9] A. V. Moro, F. S. P. Cardoso, C. R. D. Correia, Org. Lett. 2009, 11, 3642. doi: 10.1021/ol901416e.
- [10] B. Schmidt, F. Hölter, R. Berger, S. Jessel, Adv. Synth. Catal. 2010, 352, 2463. doi: 10.1002/adsc.201000493.
- [11] J. Salabert, R. M. Sebastián, A. Vallribera, A. Roglans, C. Nájera, Tetrahedron 2011, 67, 8659. doi: 10.1016/j.tet.2011.09.046.
- [12] F. de Azambuja, C. R. D. Correia, Tetrahedron Lett. 2011, 52, 42. doi: 10.1016/j.tetlet.2010.10.132.
- [13] J. G. Taylor, R. da S. Ribeiro, C. R. D. Correia, Tetrahedron Lett. 2011, 52, 3861. doi: 10.1016/j.tetlet.2011.05.039.
- [14] E. T. da Penha; J. A. Forni, A. F. P. Biajoli, C. R. D. Correia, Tetrahedron Lett. 2011, 52, 6342. doi: 10.1016/j.tetlet.2011.09.014.
- [15] R. G. Kalkhambkar, K. K. Laali, Tetrahedron Lett. 2011, 52, 1733. doi: 10.1016/j.tetlet.2011.02.021.
- [16] F. Le Callonnec, E. Fouquet, F.-X. Felpin, Org. Lett. 2011, 13, 2646. doi: 10.1021/ol200752x.
- [17] N. Oger, F. Le Callonnec, D. Jacquemin, E. Fouquet, E. Le Grognec, F.-X. Felpin, Adv. Syn. Cat. 2014, 356, 1065. doi: 10.1002/adsc.201301144.
- [18] N. Oger, E. Le Grognec, F.-X. Felpin, J. Org. Chem. 2014,79, 8255. doi: 10.1021/jo501468z.
- [19] D. Cortés-Borda, K. V. Kutonova, C. Jamet, M. E. Trusova, F. Zammattio, C. Truchet, M. Rodriguez-Zubiri, F.-X. Felpin, Org. Process Res. Dev. 2016, 20, 1979. doi: 10.1021/acs.oprd.6b00310.
- [20] D. S. Gaikwad, D. M. Pore, Synlett 2012, 23, 2631. doi: 10.1055/s-0032-1317477.
- [21] T. Nakano, M. Miyahara, T. Itoh, A. Kamimura, Eur. J. Org. Chem. 2012, 2161. doi: 10.1002/ejoc.201101703.
- [22] I. Peñafiel, I. M. Pastor, M. Yus, Eur. J. Org. Chem. 2012, 3151. doi: 10.1002/ejoc.201200181.
- [23] J. Salabert, R. M. Sebastián, A. Vallribera, J. F. Cívicos, C. Nájera, Tetrahedron 2013, 69, 2655. doi: 10.1016/j.tet.2013.01.049.
- [24] M. Gholinejad, Appl. Organomet. Chem. 2013, 27, 19. doi: 10.1002/aoc.2932.
- [25] J. Xiao, Z. Lu, Z. Li, Y. Li, Appl. Organomet. Chem. 2015, 29, 646. doi: 10.1002/aoc.3346.
- [26] T. Y. Chaudhari, A. Hossian, M. K. Manna, R. Jana, Org. Biomol. Chem. 2015,13, 4841. doi: 10.1039/C5OB00235D.
- [27] K. V. Kutonova, M. E. Trusova, A. V. Stankevich, P. S. Postnikov, V. D. Filimonov, Beilstein J. Org. Chem. 2015, 11, 358. doi: 10.3762/bjoc.11.41.
- [28] E. Mohammadi, B. Movassagh, J. Mol. Cat. A. 2016, 418-419,158.
- doi: 10.1016/j.molcata.2016.03.045.
- [29] B. Schmidt, F. Wolf, H. Brunner, Eur. J. Org. Chem. 2016, 2972. doi: 10.1002/ejoc.201600469.
- [30] W. Khodja, A. Leclair, J. Rull-Barrull, F. Zammattio, K. V. Kutonova, M. E. Trusova, F.-X. Felpin, M. Rodriguez-Zubiri, New J. Chem. 2016, 40, 8855. doi: 10.1039/C6NJ01717G.
- [31] H. Qin, Q. Zheng, G. A. L. Bare, P. Wu, K. B. Sharpless, Angew. Chem. 2016, 128, 14361. doi: 10.1002/ange.201608807.
- [32] B. Schmidt, F. Wolf, C. Ehlert, J. Org. Chem. 2016, 81, 11235. doi: 10.1021/acs.joc.6b02207.
- [33] S. Pape, L. Daukšaitė, S. Lucks, X. Gu, H. Brunner, Org. Lett. 2016, 18, 6376. doi: 10.1021/acs.orglett.6b03268.
- [34] D. S. Gaikwad, K. A. Undale, D. B. Patil, D. M. Pore, S. N. Korade, A. A. Kamble, Res. Chem. Intermed. 2017, 43, 4445. doi: 10.1007/s11164-017-2888-5.
- [35] S. Lucks, H. Brunner, Org. Process Res. Dev. 2017, 21, 1835. doi: 10.1021/acs.oprd.7b00279.
- [36] H. Brunner, L. Vedder, Chem. Cat. Chem. 2019, 11, 698. doi: 10.1002/cctc.201801519.
- [37] N. Riemer, M. Shipman, P. Wessig, B. Schmidt, J. Org. Chem. 2019, 84, 5732. doi: 10.1021/acs.joc.9b00627 .
- [38] L. Mateliene (née Dauksaite), J. Knaup, F. von Horsten, X. Gu, H. Brunner, Eur. J. Org. Chem. 2020, 127. doi: 10.1002/ejoc.201901708.
- [39] S. P. Vibhute, P. M. Mhaldar, R. V. Shejwal, G. S. Rashinkar, D. M. Pore, Tetrahedron Lett, 2020, 61, 151801. doi: 10.1016/j.tetlet.2020.151801.
反应机理
4-羟基-2-丁烯酸甲酯参与的Heck-Matsuda反应[1]-[3]
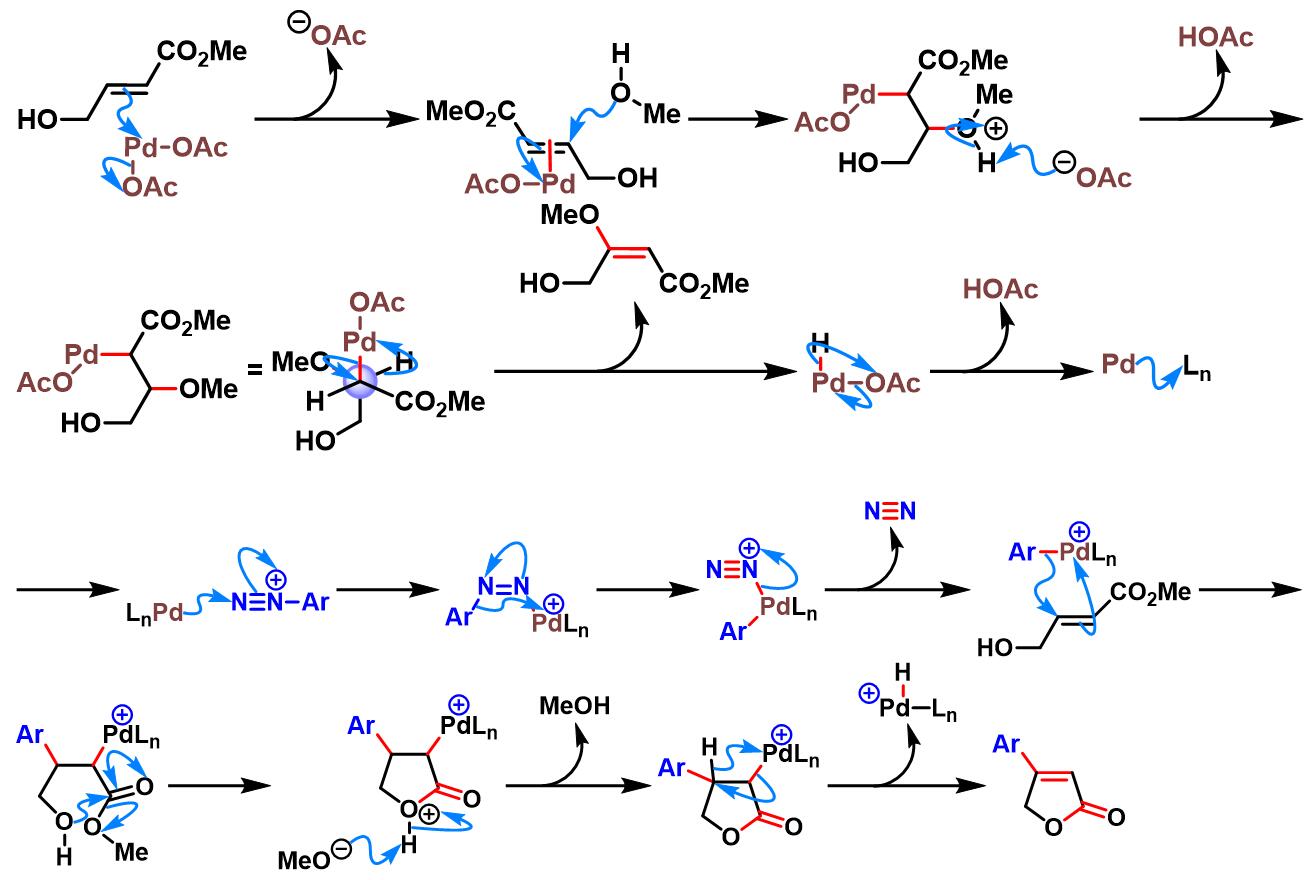
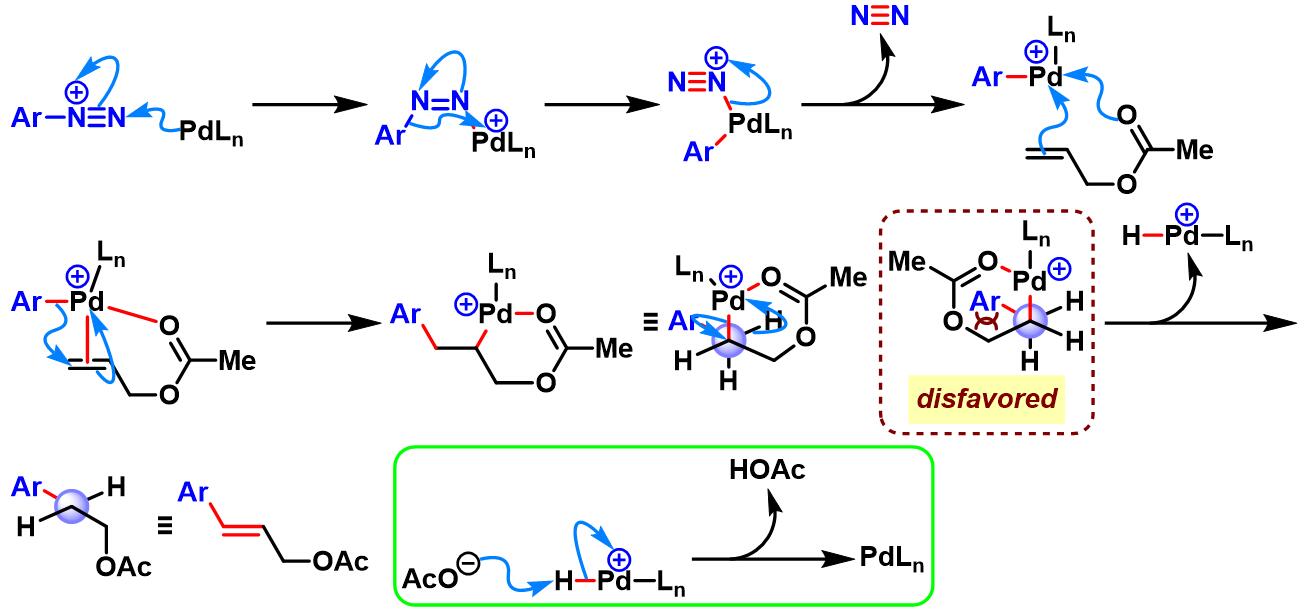
丙烯酸酯的双重芳基化[3]-[4]

烯丙酯参与的Heck-Matsuda反应[5]
参考文献
- [1] S. Cacchi, G. Fabrizi, A. Goggiamani, A. Sferrazza, Synlett 2009, 1277. doi: 10.1055/s-0028-1088132.
- [2] A. A. Sabino, A. H. L. Machado, C. R. D. Correia, M. N. Eberlin, Angew. Chem. Int. Ed. 2004, 43, 2514. doi: 10.1002/anie.200353076.
- [3] B. Schmidt, F. Wolf, H. Brunner, Eur. J. Org. Chem. 2016, 2972. doi: 10.1002/ejoc.201600469.
- [4] J. G. Taylor, R. da S. Ribeiro, C. R. D. Correia, Tetrahedron Lett. 2011, 52, 3861. doi: 10.1016/j.tetlet.2011.05.039.
- [5] V. H. M. da Silva, N. H. Morgon, C. R.D. Correia, A. A. C. Braga, J. Organomet. Chem. 2019, 896, 5. doi: 10.1016/j.jorganchem.2019.05.023.
反应实例
官能团化的benzalacetone的合成[1]
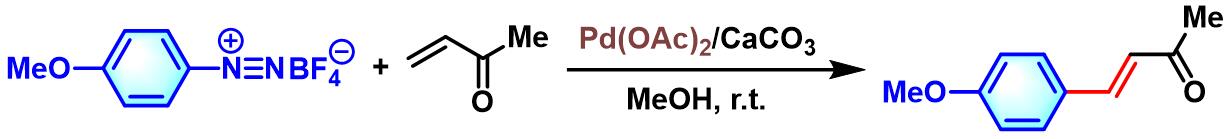
四氟亚乙基烯烃的芳基化[2]-[3]
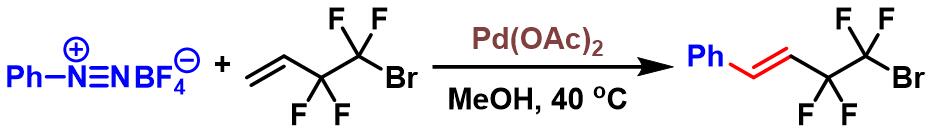
三氟甲基化多取代烯烃的合成[4]
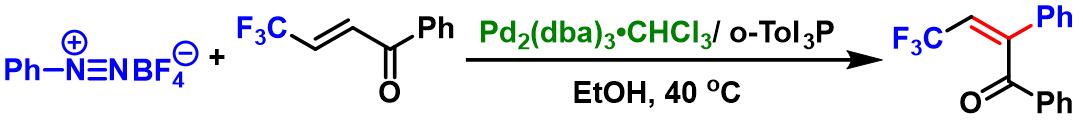
烯烃的芳基化[5]

参考文献
- [1] T. Stern, S. Rückbrod, C. Czekelius, C. Donner, H. Brunner, Adv. Synth. Catal. 2010, 352, 1983. doi: 10.1002/adsc.200900868.
- [2] Y. Sakaguchi, S. Yamada, T. Konno, T. Agou, T. Kubota, J. Org. Chem. 2017, 82, 1618. doi: 10.1021/acs.joc.6b02793.
- [3] T.Konno, S. Yamada, A. Tani, M. Nishida, T. Miyabe, T. Ishihara, J. Fluor. Chem. 2009, 130, 913. doi: 10.1016/j.jfluchem.2009.07.002.
- [4] T. Konno, S. Yamada, A. Tani, T. Miyabe, T. Ishihara, Synlett 2006, 3025. doi: 10.1055/s-2006-951496
- [5] A.S.Singh, S.S.Shendage, J.M.Nagarkar, Tetrahedron Lett. 2013, 54 ,6319. doi: 10.1016/j.tetlet.2013.09.027.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!

















No comments yet.