
导读:
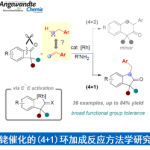
近日,上海交通大学/上海师范大学赵宝国教授团队利用自主开发的手性吡哆胺催化剂,实现了α-酮膦酸酯的仿生不对称转氨化,以良好产率和高达98% ee的对映选择性高效构建一系列手性α-氨基膦酸酯,彰显维生素B6启发催化剂在不对称合成中的强大潜力。
“ Asymmetric biomimetic transamination of α-keto phosphonates enabled by chiral pyridoxamines and synergistic solvent.
Dongchen Cai, Longjie Huang, Zhuochuan Wang, Siqi Liu*, Xiao Xiao* & Baoguo Zhao*
Nat Commun 2026. DOI:10.1038/s41467-026-69567-x
正文
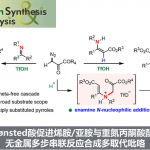
鉴于胺产物的重要性以及酮底物的易得性和低成本,因此以酮为原料转化为手性伯胺极具价值(Figure 1a)。传统化学法需先将酮转化为亚胺,再利用亚胺的反应性,如还原、亲核加成或1,3-氢迁移等反应,最后脱保护获得手性伯胺。相比之下,生物转氨——由转氨酶催化、经两步1,3-质子迁移完成——可直接生成N-未保护的手性伯胺,省去缩合、保护与脱保护步骤,因而自20世纪50年代起广受关注。如今,ω-转氨酶(ω-TAs)已实现工业化应用,成功用于多种药物的手性伯胺合成。与此同时,仿生转氨策略也快速发展:通过模拟维生素B6依赖型转氨酶的结构与机制,手性吡哆胺类催化剂兼具高催化效率、高对映选择性、反应条件温和及结构可调等优势。
手性α-氨基膦酸酯是一类重要的有机磷化合物,广泛应用于药物化学与合成化学。且其独特结构赋予多种生物活性:如抗菌药磷霉素衍生物Alafosfalin、hFPPs抑制剂、阿尔茨海默病治疗中咪唑啉I2受体(I2-IRs)配体,以及基质金属蛋白酶(MMPs)抑制剂(Figure 1b)。鉴于其药理潜力、结构多样性和合成适应性,科学家开发了多种合成策略来制备手性α-氨基膦酸酯。如Kabachnik–Fields 反应、Pudovik 反应、亚胺膦酸酯的亲核加成或亲核加成(Figure 1c,上)。近年来,仿生1,3-质子迁移策略备受关注,开发了一系列构建手性α-氨基膦酸酯的方法(Figure1c方框)。此外,尽管酶促转氨效率最高,但天然转氨酶通常难以识别非蛋白源α-酮膦酸酯,受限于固有底物特异性。因此,仿生不对称转氨成为制备非天然手性α-氨基膦酸酯的重要替代路径。然而,能直接、高效、普适地获得N-未保护产物的方法仍十分稀缺。
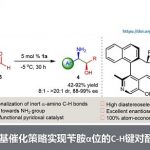
上海交通大学/上海师范大学的赵宝国教授基于辅酶 (coenzyme)Vitamin B6的相关结构,设计出一系列新型的手性吡哆胺 (pyridoxamine)催化体系,并顺利实现了一系列α-酮酸/α-酮酰胺的仿生转氨化(Org. Lett. 2015, 17, 5784;Org. Lett. 2016, 18, 3658;J. Am. Chem. Soc. 2016, 138, 10730.;Nat. Commun. 2021, 12, 5174)。近日,该团队利用自主开发的手性吡哆胺催化剂,实现了α–酮膦酸酯的仿生不对称转氨化。该反应条件温和,产率高达86%,对映选择性达98%(Figure 1d),适用于结构多样的底物。机理研究表明,催化剂精细结构与溶剂微环境共同决定立体选择性。
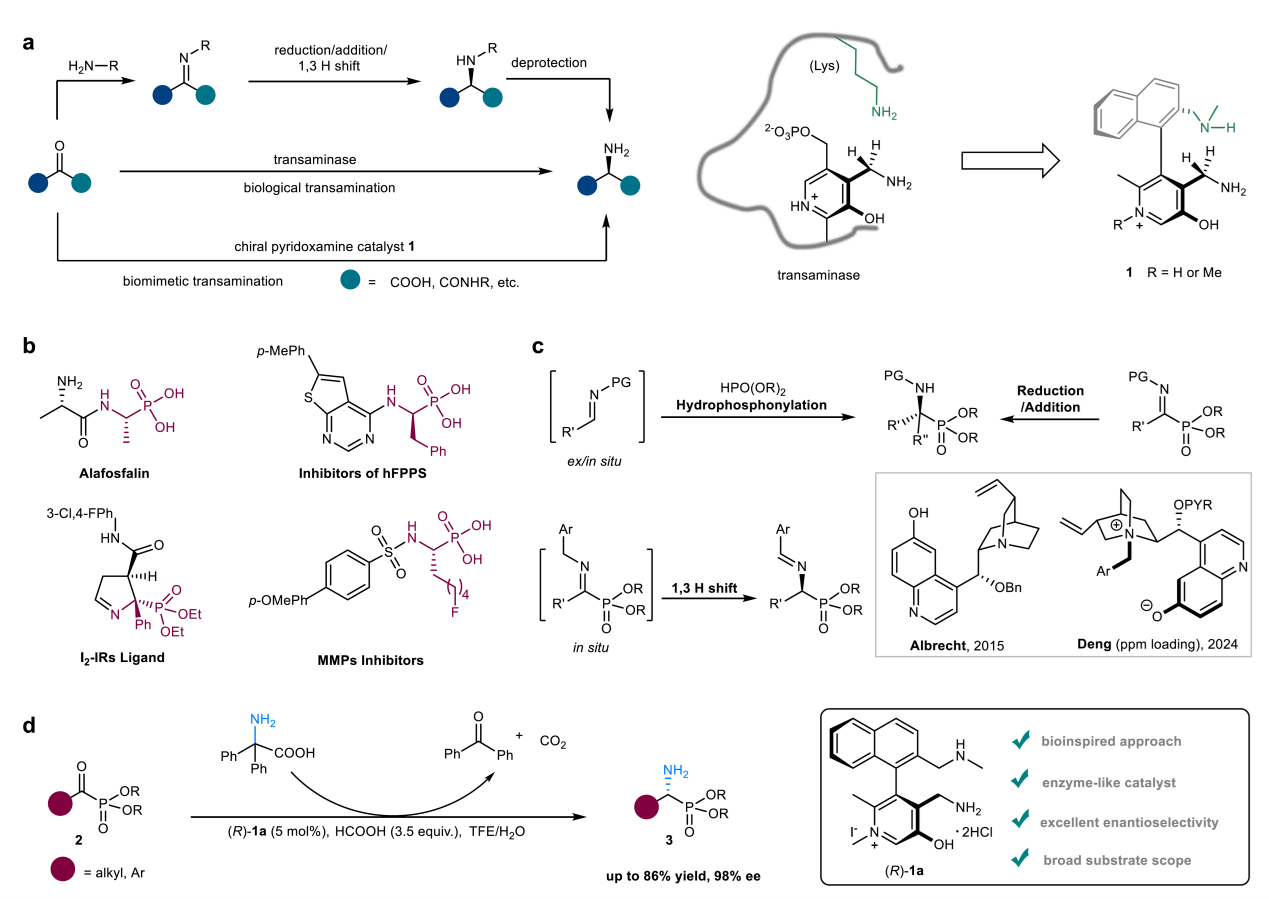
Fig. 1 Asymmetric biomimetic transamination of α-keto phosphonates enabled by chiral pyridoxamine catalysts.
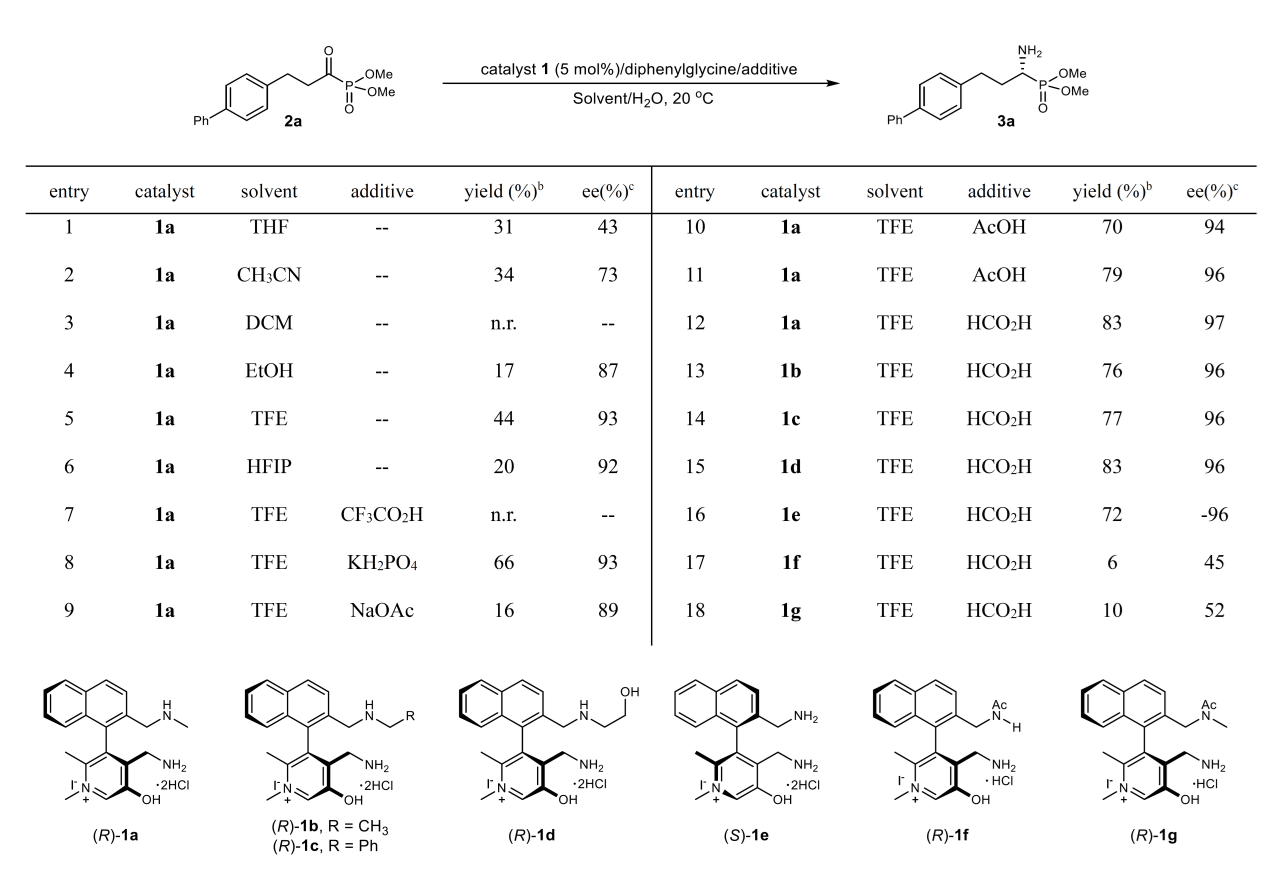
Table 1 Reaction conditions optimization
首先,作者以α-酮膦酸酯2a作为模型底物,以二苯甘氨酸(4a)为氮源、手性吡哆胺1a为催化剂对反应条件进行了筛选。研究结果表明:醇类溶剂更利于提升对映选择性,其中三氟乙醇(TFE)效果最优;酯基适配性良好:甲酯与异丙酯均可高效反应; HCOOH作为添加剂可显著加速反应,以83%产率和97% ee获得产物(Table 1,enrtires 12);而CF₃COOH(entry 7)或NaOAc(entry 9)则大幅降低产率与对映选择性。结果表明,该仿生转氨反应在中性至弱酸性条件下表现最佳,与生物转氨体系一致。多种替代氨基供体(N,N-二甲基甘氨酸4b、甲基苯基甘氨酸4c、苯丙氨酸4d、苄胺4e)在最优条件下产率均低于5%,因其难以驱动催化循环中吡哆醛向吡哆胺的转化;而二苯甘氨酸易脱羧,可高效再生活性催化剂。催化剂结构至关重要:含仲胺侧链的1a–e均给出高对映选择性(entries 13–16),说明1d中羟基不参与氢键催化(entry 15);伯胺催化剂1e活性略低(entry 16);酰胺型催化剂1f/1g则产率与ee值均急剧下降(entries 17–18),证实胺侧链需具备足够碱性,仲胺为催化所必需。尽管α-酮膦酸酯因膦酸酯基具离去能力可能引发酰化副反应,但在优化的弱酸性条件下未观察到此类副产物,表明酸性环境有效抑制该路径。
在最优条件下,作者对底物的兼容性进行了研究(Figure 2)。苄基与苯乙基取代的α-酮膦酸酯均顺利反应,以高对映选择性(92–98% ee)生成对应α-氨基膦酸酯3b–d。一系列含有不同取代基的苯乙基底物均可兼容(3e–n),如供电子基(3e, f, g, n)和缺电子基(3h–m)。异丙基膦酸酯(3f, 3h)、甲基膦酸酯(3g, 3i)、萘乙基(3q)与噻吩乙基(3r)、脂肪族α-酮膦酸酯(3t–af)等底物均可兼容。多种官能团(杂原子如3u、3ac;酯基, 如3v、3z;双键, 如3aa;炔键,如3ab;脂环如3ad)均耐受。含复杂结构的底物也可兼容,如香茅醛和脱氧胆酸衍生的α-酮膦酸酯(3ae, 3af)雄激素受体拮抗剂衍生的底物3ag等均可兼容。值得注意的是,多个产物与天然氨基酸结构对应:3b(苯丙氨酸)、3c(酪氨酸)、3t(丙氨酸)、3u(甲硫氨酸)、3v(谷氨酸),表明该方法适用于构建蛋白质源氨基酸的氨基膦酸酯类似物。异丙基及缬氨酸衍生的α-酮膦酸酯则不反应,可能源于β-位阻阻碍催化循环。
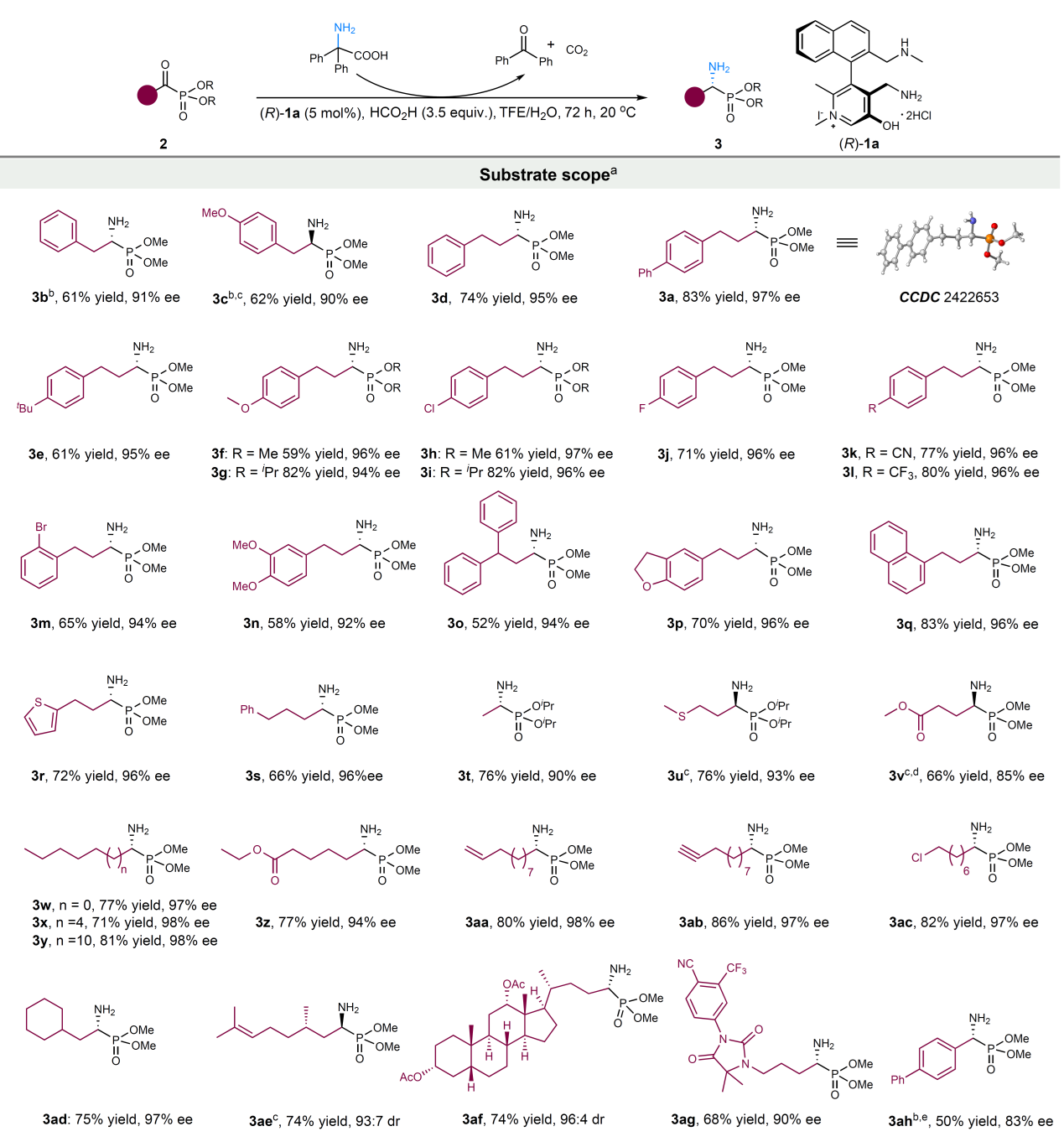
Fig. 2 Substrate scope study
随后,作者对该反应的实用性进行了研究(Figure 3)。即1)(S)-1a催化α-酮膦酸酯2a(3.0 mmol规模)转氨,以80%产率和97% ee获得(S)-3a,与小试结果(83% yield,97% ee)一致。(S)-3a经水解以96%产率转化为手性α-氨基膦酸5(Figure 3a);2)6-氟己酰膦酸酯(2ai)在最优条件下以71%产率和96%ee获得3ai,其经磺酰氯6保护氨基(75% yield,95% ee),再水解即得MMPs抑制剂8(Figure 3b);3)伊沙佐米对映体类似物12快速构建:2aj经(S)-1a催化转氨得3aj(60% yield,92% ee),再与(2,5-二氯苯甲酰基)甘氨酸(10)缩合,经TMSBr水解,最终以92% ee获得12(Figure 3c)。
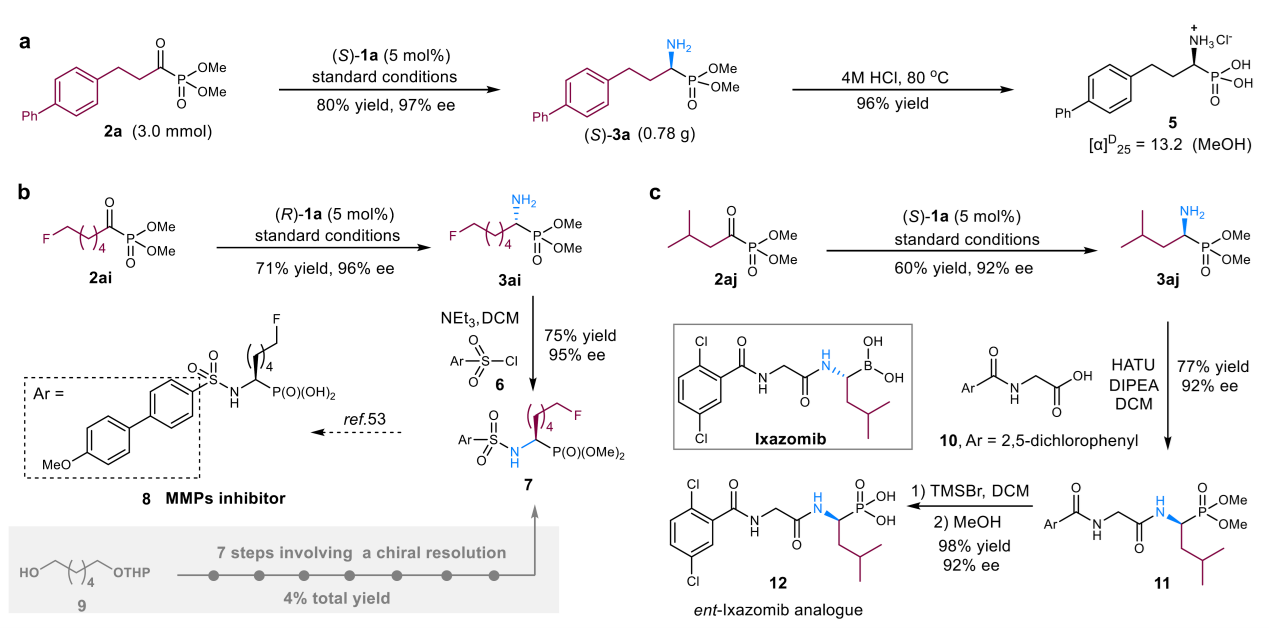
Fig .3 Derivatization of α-aminophosphonates
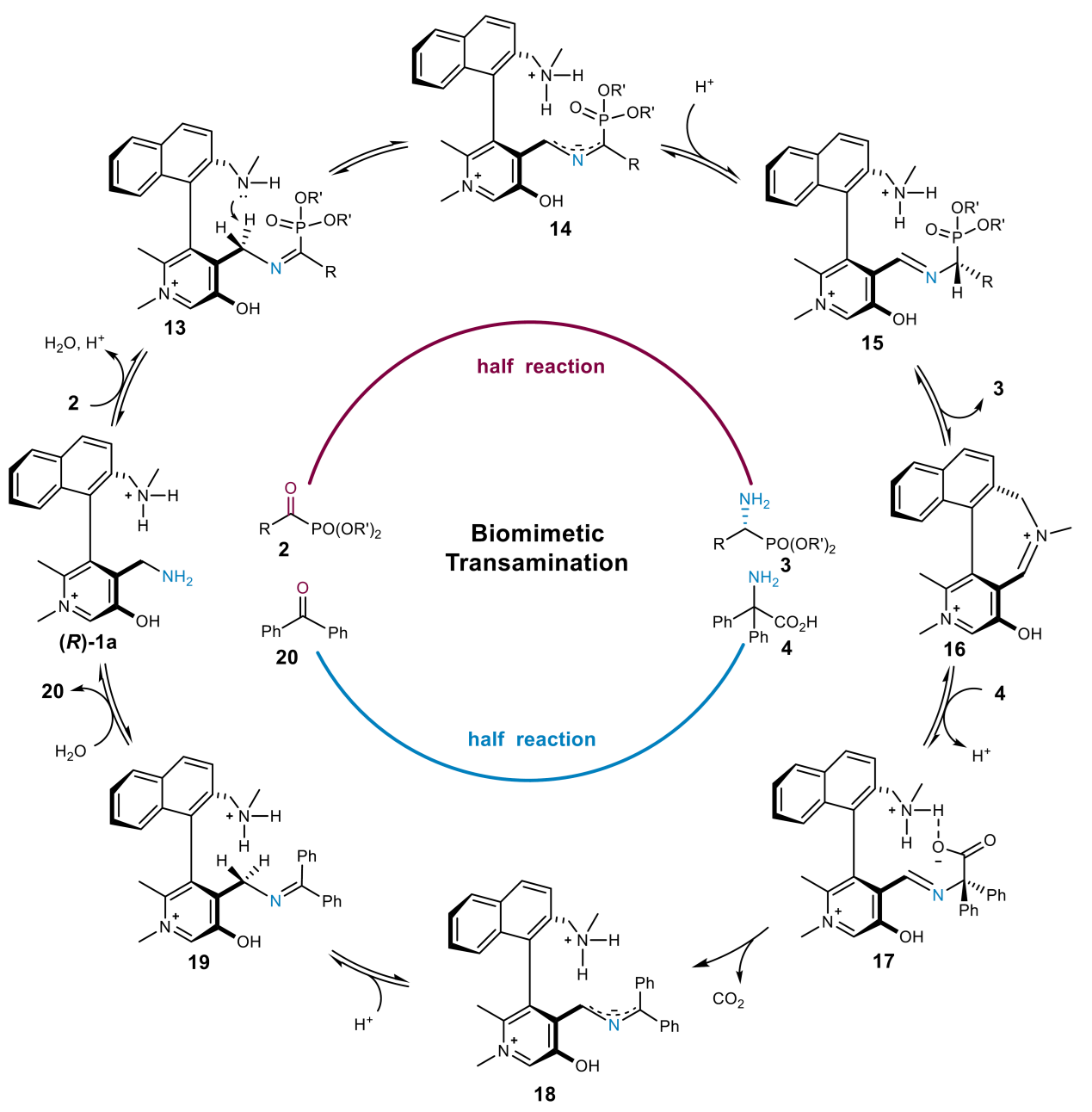
Fig. 4 Proposed mechanism for transamination of α-keto phosphonates
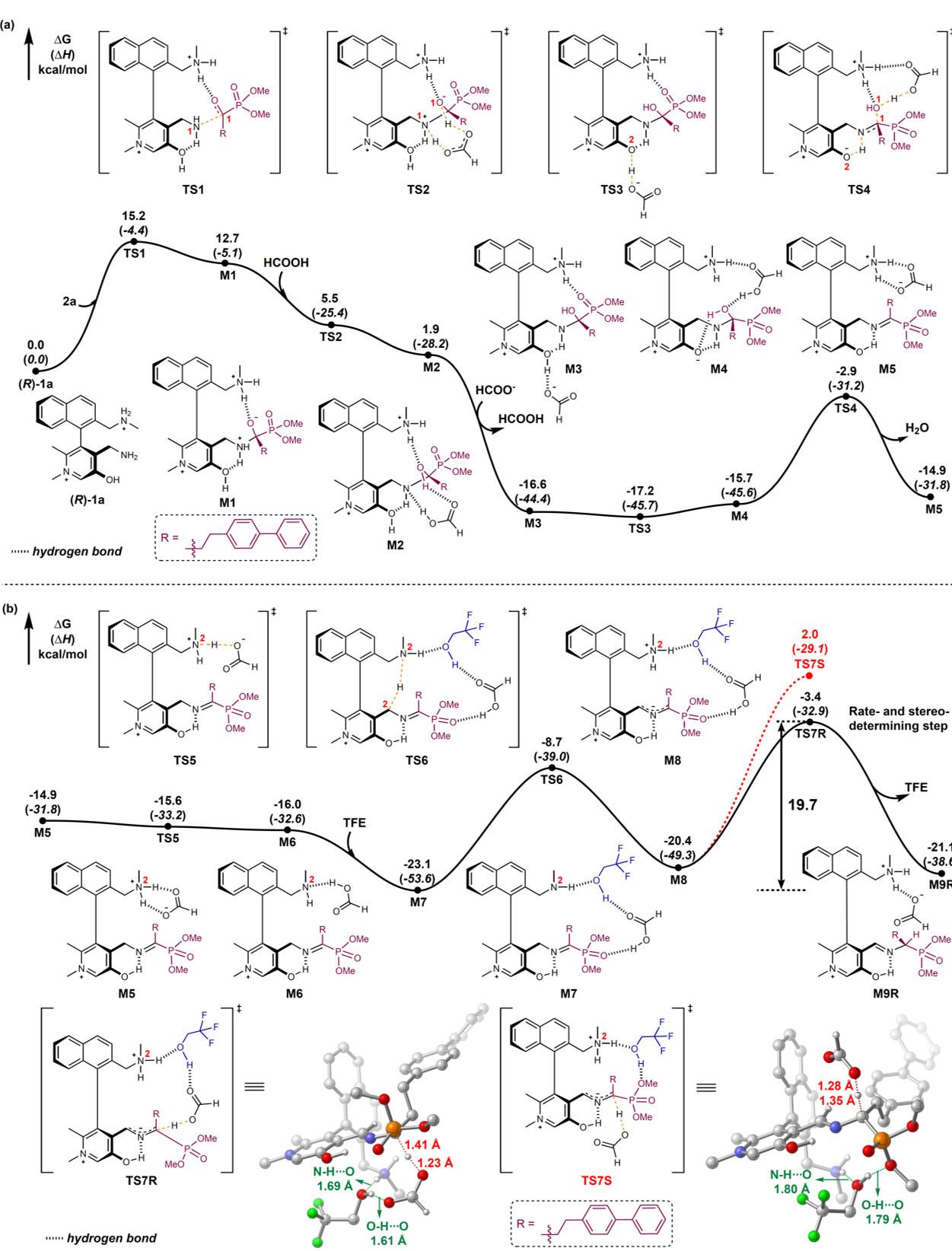
Fig .5 Energy profiles and geometries of key transition states for the transamination of α-keto phosphonate.
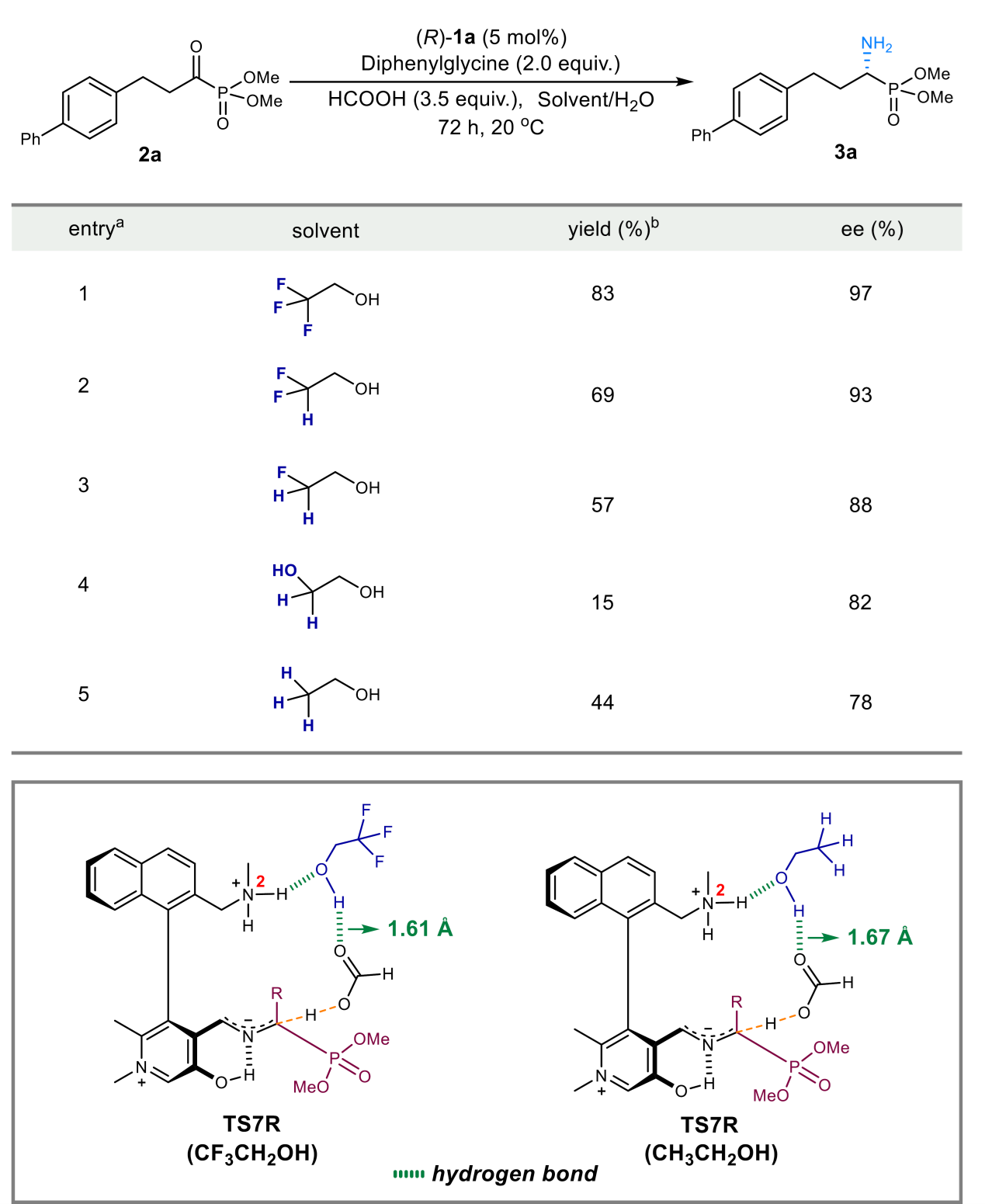
Fig. 6 Control experiments on alcohol solvents and TS7R transition states in TFE and EtOH.
其次,作者对该反应的机理进行了研究(Figure 4-6)。即DFT计算表明(Figure 5):(R)-1a中的胺基亲核进攻2a,经TS1(ΔG‡ = 15.2 kcal/mol)生成M1;甲酸氢键网络协助下,质子经TS2从N1转移至O1,得M2;随后发生配体交换(甲酸解离、甲酸根与酚羟基配位),生成M3;经TS3质子由酚羟基转移至甲酸根,得M4;M4经C1–O1断裂与水消除(TS4)生成M5;再经TS5(N2→甲酸根质子转移)得M6;TFE进入并与侧链N2–H氢键结合,形成M7;侧链胺经TS6(ΔG‡ = 14.4 kcal/mol)对苄位C2–H脱质子,生成M8;最后,立体选择性质子化经TS7R(ΔG‡ = 19.7 kcal/mol)生成手性M9R,释放(R)-3a。竞争路径TS7S能垒更高(ΔG‡ = 25.1 kcal/mol),几何分析表明TS7R中N–H···O与O–H···O氢键距离更短,氢键网络稳定作用更强,故R路径具显著动力学优势。综上,TS7R质子化是首步半转氨的决速步与立体决定步。另外一方面,三氟乙醇在不对1,3-质子迁移过程中的作用研究表明(Figure 5):β位吸电子效应对立体诱导至关重要。DFT进一步显示:乙醇中TS7R/TS7S能垒差小于TFE中,与实验ee降低趋势一致。且TFE中TS7R的氢键更强(酸性溶剂增强甲酸根质子供体能力),从而提升对映选择性与催化活性;弱酸性醇则导致立体控制与效率下降。因此,TFE对实现高活性与高对映选择性具有决定性作用。
综上,作者提出了可能的反应机理(Figure 4)。即该反应遵循典型的吡哆胺催化转氨机制,模拟酶促转氨的两个半反应:第一半反应中,α-酮膦酸酯2与手性吡哆胺(R)-1a缩合形成席夫碱13,其1,3-质子迁移由催化剂内胺侧链促进;第二半反应中,二苯甘氨酸(4a)的氨基经席夫碱17/19异构化转移至内亚胺16,再生(R)-1a(图4)。尽管中间体14或18理论上可作为质子供体,但在含HCOOH、CF₃CH₂OH和H₂O的反应体系中,外部质子源介导或质子穿梭途径亦属合理。
总结
上海交通大学/上海师范大学赵宝国教授团队利用自主开发的手性吡哆胺催化剂,实现了α-酮膦酸酯的仿生不对称转氨化,以良好产率和高达98% ee的对映选择性高效构建一系列手性α-氨基膦酸酯,彰显维生素B6启发催化剂在不对称合成中的强大潜力。该方法优势显著:直接引入N-未保护氨基、高原子经济性、宽底物适用范围及优异的复杂分子兼容性。DFT支持的机理研究阐明,立体选择性源于三氟乙醇协同强化的氢键网络对关键过渡态的稳定作用,且1,3-质子迁移为决速步。此溶剂–催化剂协同机制精准复现了酶催化中的固有协同原理。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
关注Chem-Station抖音号:79473891841
请登陆TCI试剂官网查看更多内容
https://www.tcichemicals.com/CN/zh/














No comments yet.