

摘要:四川大学余达刚教授课题组(主页)最近报道了首例在可见光作用下,Pd催化剂可在温和条件下实现C(sp3)-H键与非活泼烷基溴化物的直接交叉偶联反应,反应底物范围广,官能团兼容性好。机理研究表明,Pd络合物作为反应的光催化剂,涉及自由基过程。
http://onlinelibrary.wiley.com/doi/10.1002/anie.201704513/abstractDOI:10.1002/anie.201704513
一、研究背景
过去的几十年里,可见光氧化还原催化作为一个有用的方法在温和条件下实现了新颖的有机转化[1]。除了有机染料、Ru和Ir络合物外,化学家们将许多其他过渡金属络合物作为光催化剂用于有机合成(图1,A)[2-3]。尽管Pd催化剂已经广泛用于交叉偶联反应并且它局限的光物理性质也得到了研究,然而将简单易得无需外部光敏剂的Pd络合物用于可见光氧化还原过程却鲜有报道[4]。
Pd催化的C-H键直接官能团化是有机化学中非常有用且吸引人的方法[5]。虽然已经取得巨大进展,然而Pd催化的非活泼烷基卤,尤其是3°烷基卤化物参与的C-H键官能团化却相对滞后,主要挑战在于迟缓的氧化加成和还原消除步骤以及诸如 -H消除和烷基钯中间体的质子化这些竞争性副反应的存在[6]。最近,Fu、Zhou和Alexanian三个研究组各自独立实现了Pd催化的(杂)芳烃和非活泼烷基卤化物的自由基烷基化反应[7],然而Pd催化的C(sp3)-H键与非活泼3°和2°烷基卤的烷基化反应仍未被探索。研究人员设想能否通过引入可见光作为能量来源去活化Pd催化剂实现自由基交叉偶联来解决上述挑战。作为课题组在可见光驱动过渡金属催化作用兴趣的延续,文章报道了一个独特的可见光氧化还原催化体系,Pd(PPh3)4作为仅有的催化剂,实现了首例N-芳基四氢异喹啉分子中C(sp3)-H键与非活泼烷基卤化物的交叉偶联反应。(图1,B)此外,(杂)芳基C(sp2)-H键的烷基化在温和条件下也成功实现。
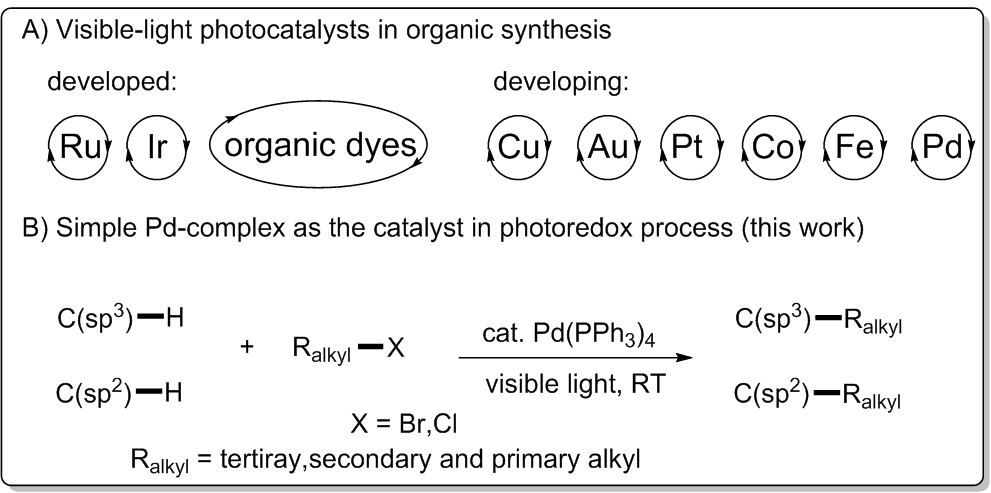
图1可见光催化
二、最佳反应条件优化
根据设想,研究人员开始探究2-苯基-1,2,3,4-四氢异喹啉(1)和叔丁基溴(2)的反应。通过筛选诸如溶剂、碱和Pd催化剂等多个变量,研究人员欣喜地发现以Pd(PPh3)4为催化剂,无需任何外加的光引发剂,可以90%的产率得到预期产物(3)(表1,entry 1),对照实验表明磷配体、Pd催化剂以及可见光辐射都是反应必需的(entries 2-8)。重要的是,没有光存在时的热反应并不能得到预期产物(entry 9)。
表 1反应条件的筛选

三、底物拓展
以上述观察到的结果为基础,研究人员首先探究了非活泼3°烷基卤的普适性(图2,sectionA),许多大位阻的底物可以很方便地转化为需要的产物,产率从中等到优秀不等,证实了该催化体系的高适用性。值得一提的是,利用光催化策略,在没有检测到异构化产物的情况下,这些“拥挤”的季碳中心可以高效形成。包含羰基官能团的底物(9和16)也可顺利进行反应。除了3°烷基溴,多种不活泼的2°和1°烷基溴底物也得到了探究(图2,sectionB和C)。不难看出,2°烷基溴(20–28),包括不同大小的环状底物和非环状底物均可完美参与反应;此外,一系列非活泼的1°烷基溴也能很好达到预期目标(29–42)。值得注意的是,LiBr对于促进苄氯的反应很有帮助,预示着苄溴是反应的关键偶联部分(40–42)。此反应兼容许多官能团,包括羟基(33)、酯基(34和35)、醚键(36和37)以及氯原子(38和42)、氟原子(41),这些底物对于随后的转化反应很有利。
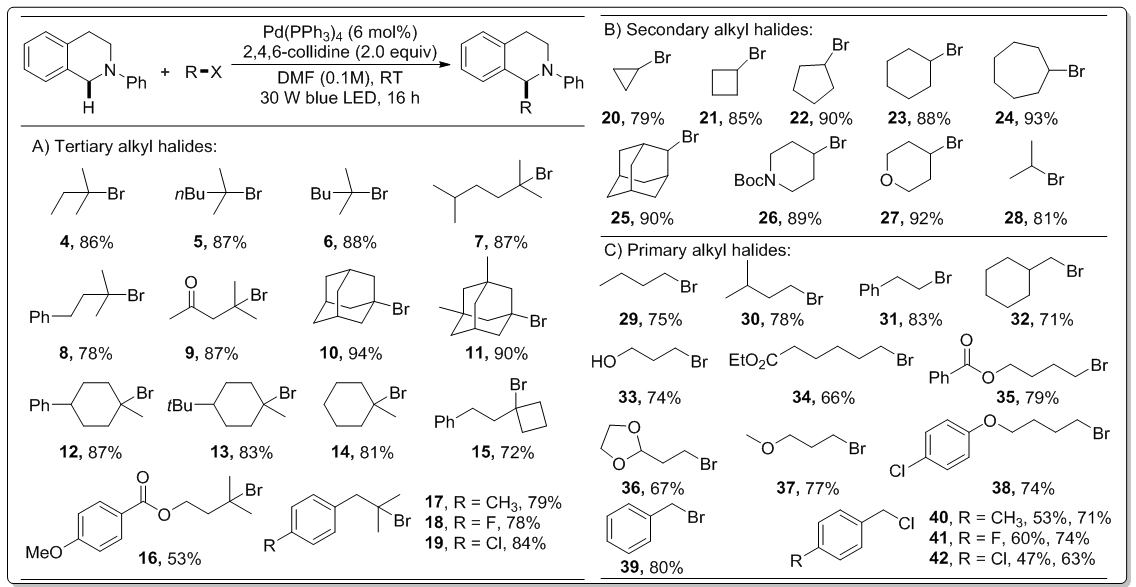
图 2底物烷基卤的适用范围
该催化体系对于另一底物N-芳基四氢异喹啉也具有普遍性(图3)。与N相连的苯环的邻、间、对位上被氟、氯、溴原子、甲基、甲氧基取代都可以得到对应的烷基化产物(43–54),产率可观。四氢异喹啉的苯环上进行取代,例如双甲氧基取代的底物(55)可以较高的产率81%得到烷基化产物。
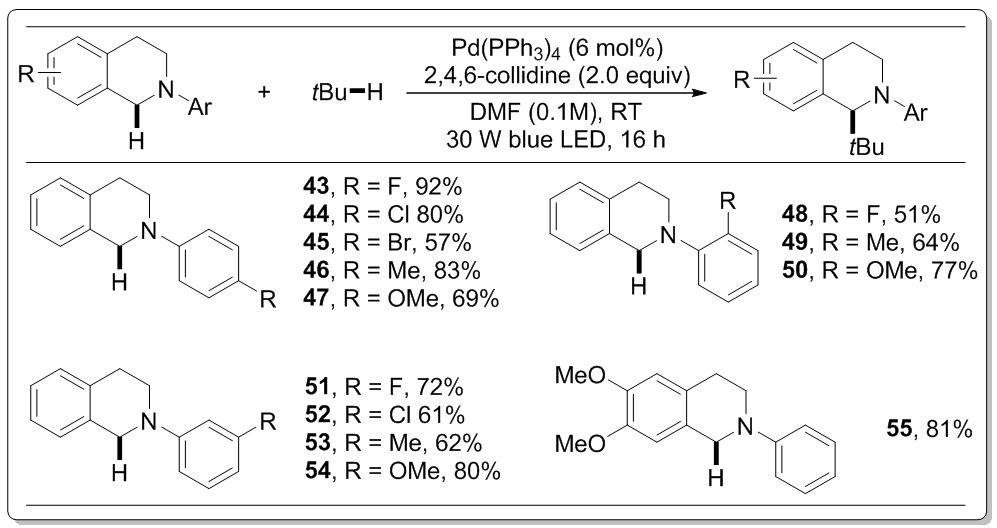
图 3底物N-芳基四氢异喹啉的适用范围
鉴于芳杂环化合物官能团化的重要性,研究人员在温和条件下将该催化体系用于吲哚的分子内C(sp2)-H键的烷基化反应。仅对碱和溶剂作出小的改变,环化反应即可顺利进行,产率中等偏上(图4)。吲哚环上取代基的电性对反应影响不大,C3位上的吸电子基对产率有一定的提高(58和59)。此方法提供了一个温和且安全的吲哚官能化方法,避免利用有机锡烷和紫外光。此外,该光催化方法还可以运用到芳杂环化合物的分子间C-H键烷基化反应(图 5)。苯并噁唑(64)、呋喃(65)、噻吩(66–68)和2-甲基吡啶-N-氧化物(69)都可以和溴代金刚烷反应,产物包含了一个很难得到的季碳中心。在上述例子中,都得到了C2位选择性烷基化的产物,说明吸电子基的存在对于反应活性和区域选择性很重要。
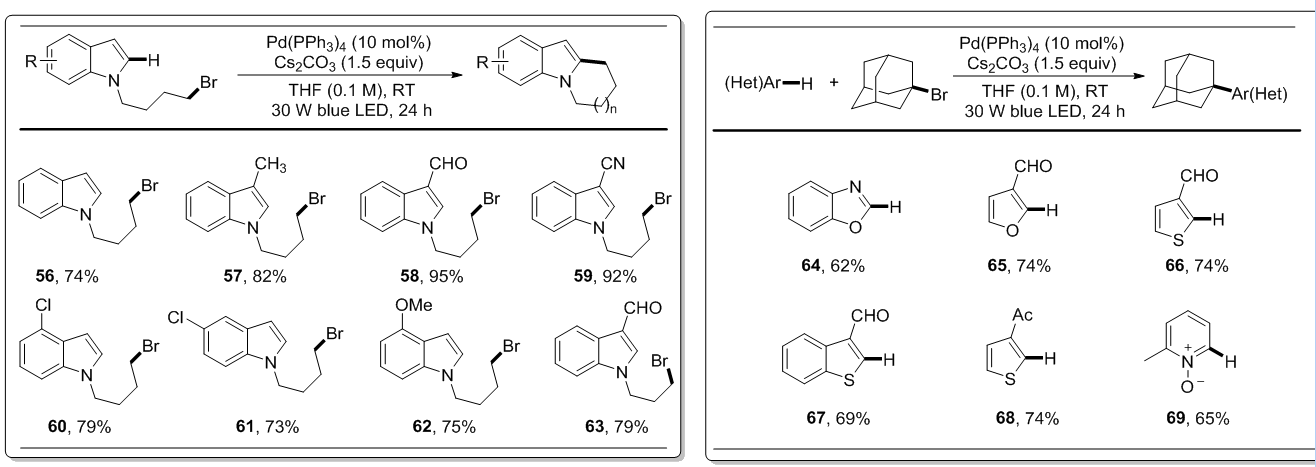
图 4吲哚分子内环化反应 图 5杂芳香烃的分子间交叉偶联
四、机理研究
为了对反应有更深入的理解,机理研究主要从自由基捕获实验、竞争反应以及radical-clock反应展开,上述实验揭示此反应是通过自由基机理进行的。为了证实Pd(0)是吸收光的物质,研究人员进行了光物理性质的探究,包括吸收和发射光谱。荧光猝灭实验和Stern-Volmer研究揭示烷基溴化物猝灭了激发态Pd(PPh3)4,它可能是参与了激发态的Pd(0)络合物的单电子转移过程(single-electrontransfer,SET)。在已获得的结果和前有报道的基础上,研究人员提出来底物1和底物2反应的可能机理(图6,A)。在可见光辐射下,活泼的Pd(0)络合物A与底物2经历一个SET过程,产生一个叔丁基自由基C和Pd(I)络合物B,随后B与另一底物1作用产生一个自由基正离子D,自身复原并继续参与催化循环。自由基正离子D在碱的作用下去质子生成 -氨基自由基E,最后与自由基C偶联得到最终产物。对于杂环化合物烷基化反应的机理(图6,B),由激发态的Pd0Ln*和烷基卤化物的SET过程开始,产生一个烷基自由基和PdILnBr络合物。随后碳自由基加成到杂环上形成一个环状的自由基中间体,再经历SET、去质子和重新芳构化过程得到产物。

图 6可能的反应机理
五、结论
总结起来,课题组发展了一个独特的可见光促进的Pd催化过程,可在温和条件下实现C-H键的高效自由基烷基化反应,一系列非活泼的烷基卤化物可以中等到优秀的产率形成C(sp3)-C(sp3)和C(sp2)-C(sp3)键,该反应底物范围广(> 60个底物例子),官能团兼容性好,可温和地构建季碳中心。
参考文献
[1]C. K. Prier, D. A. Rankic, D. W. C.MacMillan, Chem. Rev. 2013, 113, 5322. DOI: 10.1021/cr300503r [2]For leading reviews with copper complexes or a gold complex asphotocatalysts, see: a) S. Paria, O. Reiser, ChemCatChem 2014, 6, 2477. DOI: 10.1002/cctc.201402237; b) A. C. Hernandez-Perez, S. K. Collins, Acc. Chem. Res. 2016, 49, 1557. DOI: 10.1021/acs.accounts.6b00250 [3]For selected examples of gold, platinum, cobalt, and ironcomplexes as photocatalysts, see: a) L. M. Kreis, S. Krautwald,N. Pfeiffer, R. E. Martin, E. M. Carreira, Org. Lett. 2013, 15,1634. DOI: 10.1021/ol400410m; b) J.-J. Zhong, Q.-Y. Meng, G.-X. Wang, Q. Liu, B. Chen,K. Feng, C.-H. Tung, L.-Z. Wu, Chem. Eur. J. 2013, 19, 6443. DOI: 10.1002/chem.201204572 [4]For selected palladium-catalyzed processes employing exogenous photosensitizers with visible-light irradiation, see: a) D.Kalyani, K. B. McMurtrey, S. R. Neufeldt, M. S. Sanford, J. Am.Chem. Soc. 2011, 133, 18566. DOI: 10.1021/ja208068w; b) J. Zoller, D. C. Fabry, M. A.Ronge, M. Rueping, Angew. Chem. Int. Ed. 2014, 53, 13264. DOI: 10.1002/anie.201405478 [5]B.-J. Li, S.-D. Yang, Z.-J. Shi, Synlett. 2008, 949. DOI: 10.1055/s-2008-1042907 [6]For selected reviews, see: a) A. C. Frisch, M. Beller, Angew. Chem. Int. Ed. 2005, 44, 674. DOI: 10.1002/anie.200461432; b) R.Jana, T. P. Pathak, M. S. Sigman, Chem. Rev. 2011, 111, 1417. DOI: 10.1021/cr100327p [7]a) B. Xiao, Z. J. Liu, L. Liu, Y. Fu, J. Am. Chem. Soc. 2013, 135,616. DOI: 10.1021/ja3113752; b) X. Wu, J. W. T. See, K. Xu, H. Hirao, J. Roger, J.-C.Hierso, J. Zhou, Angew. Chem. Int. Ed. 2014, 53, 13573. DOI: 10.1002/anie.201408355; c) A. R. O. Venning, P. T. Bohan, E. J.Alexanian, J. Am. Chem. Soc. 2015, 137, 3731. DOI:10.1021/jacs.5b01365.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!













No comments yet.