
作者:石油醚
引言
PI3K/Akt/mTOR 通路在多种癌症中常因突变而异常激活。其中,PI3Kα 是实体瘤中最常突变的激酶之一。因此,靶向 PI3Kα 成为关键治疗策略;选择性 PI3Kα 抑制剂在 PIK3CA 突变(编码 p110α)的 HR+/HER2− 晚期或转移性乳腺癌中展现出明确的抗肿瘤活性。目前,仅有alpelisib 和inavolisib两款选择性 PI3Kα 抑制剂获 FDA 批准用于该适应症。
XJTU-L453:前世今生
XJTU-L453 是西安交通大学李义平教授团队自主研发的新型高选择性 PI3Kα 抑制剂。其在体外和体内均具强效抑制活性,且口服生物利用度高、药代动力学性质优良,已进入临床前开发阶段。
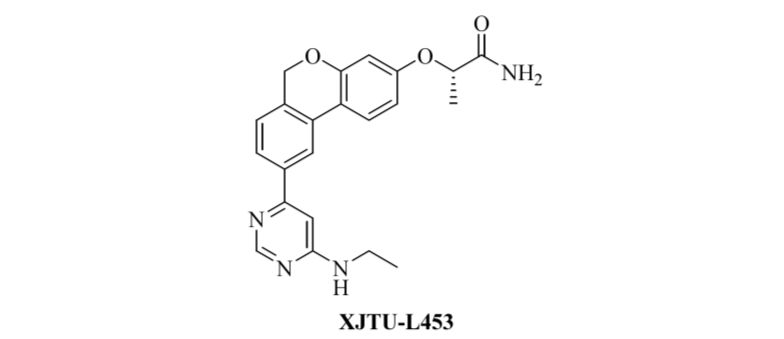
图1 PI3Kα 抑制剂- XJTU-L453
XJTU-L453的合成路线
[药物发现阶段-第一代路线]
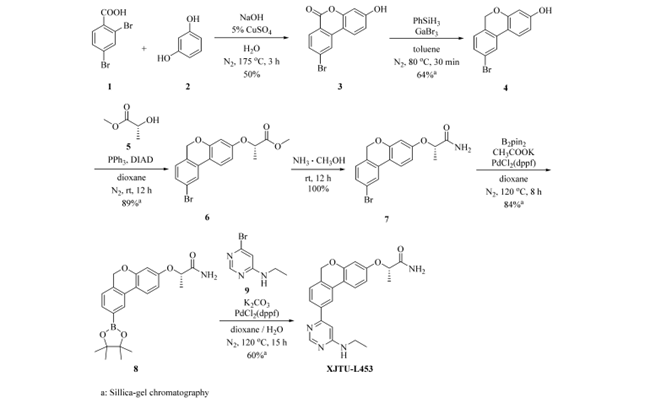
图2 XJTU-L453的第一代路线
XJTU-L453 的药物发现阶段的合成路线,如图2所示。即2,4-二溴苯甲酸(1)和间苯二酚(2)发生Hurtley reaction生成6H-benzo[c]chromen-6-one (3);(3)在苯基硅烷/GaBr3还原体系下发生还原获得6H-benzo[c]chromene (4);(4)与D-乳酸甲酯(5)发生Mitsunobu 反应获得(6);(6)随后发生氨解生成(7);(7)在钯催化的作用下与联硼酸频那醇酯反应生成(8)。最后,(8)与(9)发生Suzuki反应获得XJTU-L453。上述路线共六步,总收率仅 14%,且需五次硅胶柱层析。主要问题包括:6H-benzo[c]chromen环构建收率低且需高温;内酯还原使用吸湿性强的 GaBr₃,操作困难;Mitsunobu 反应虽收率达 89%,但原子经济性差,产生等摩尔三苯基氧膦和偶氮二甲酸酯副产物,仍需柱层析纯化。
[XJTU-L453临床阶段-第二代路线]
由于第一代路线的缺陷无法满足临床前研究对质量与批量的需求。为此,我们重新设计了合成工艺(图3),实现了 XJTU-L453 的可放大、稳健、安全制备。
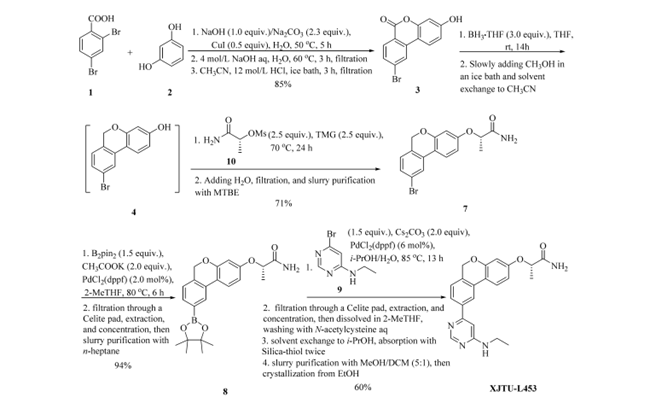
图3 XJTU-L453的第二代路线
以下是具体优化内容
1)6H-Benzo[c]chromen-6-one 3的工艺优化
基于文献,CuI/Na₂CO₃ 体系显著可提升了 Hurtley 反应产率(目标物不同)。据此,系统筛选 2,4-二溴苯甲酸(1)与间苯二酚(2)的反应条件(表1)。CuI /Na₂CO₃ 组合给出最高产率(84%,表 1,entry 1);K₂CO₃ 效果相当(84%,entry 2)。其他碳酸盐(Cs₂CO₃、Li₂CO₃、NaHCO₃;entries 3–5)或强碱(NaOH、KOH、LiOH·H₂O;entries 6–8)均导致产率下降。因分子量更低、成本更优,优选 Na₂CO₃ 而非 K₂CO₃。在 Na₂CO₃ 条件下进一步筛选铜源,发现 CuSO₄、CuCl₂、CuBr 和 Cu(OAc)₂ 均不如 CuI(entries 9–12)。最终确定 CuI/Na₂CO₃ 为最优催化体系,与文献报道一致。
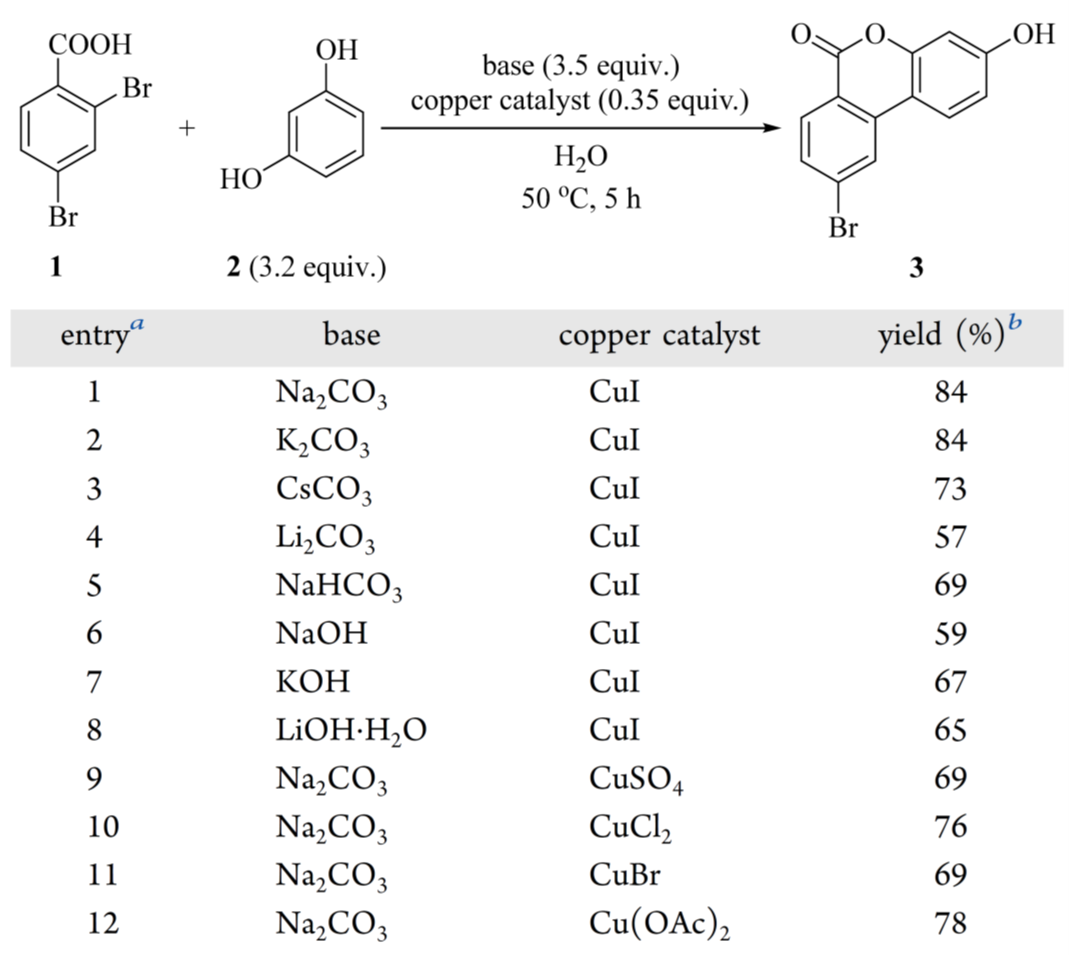
表1 Hurtley 反应条件筛选
进一步的研究集中在优化试剂用量、温度和反应时间(表 2)。在固定间苯二酚 2 与碳酸钠的摩尔比为 1:1.1 的条件下,将 2 的当量从 3.2 降至 2.5 时,产率降至 76%(表 2,entry 1)。使用 3.0 或 3.5 eq.时,产率与初始的 3.2 eq.相当(表 2,entires 2 – 4)。偏离 2 与碳酸钠 1:1.1 的摩尔比也会降低产率(表 2,entries 2、5 和 6)。对碘化铜负载量的筛选表明,较低的当量会导致产率不佳(表 2,entries 2 和 7),而将负载量增加到 0.5 eq.时,产率提高到 95%(表 2,entry 8),与初始条件相比提高了超过 10%。在该最优碘化铜负载量(0.5 eq.)下,反应温度(表 2,entries 8、9 和 10)和时间(表 2,entries 8、11 和 12)的变化所得到的结果与 50°C 反应 5 小时的条件相当。在将 2,4-二溴苯甲酸 1 与碳酸钠混合时观察到明显的泡沫问题,这给放大生产带来了挑战。这通过首先用 1.00 当量的氢氧化钠预处理 1 将其转化为钠羧酸盐,然后加入 2.30 eq的碳酸钠来缓解。这种改进抑制了泡沫的产生,并以 94% 的产率得到 3(表 2,entry13)。因此,选择第entry13的条件进行放大。
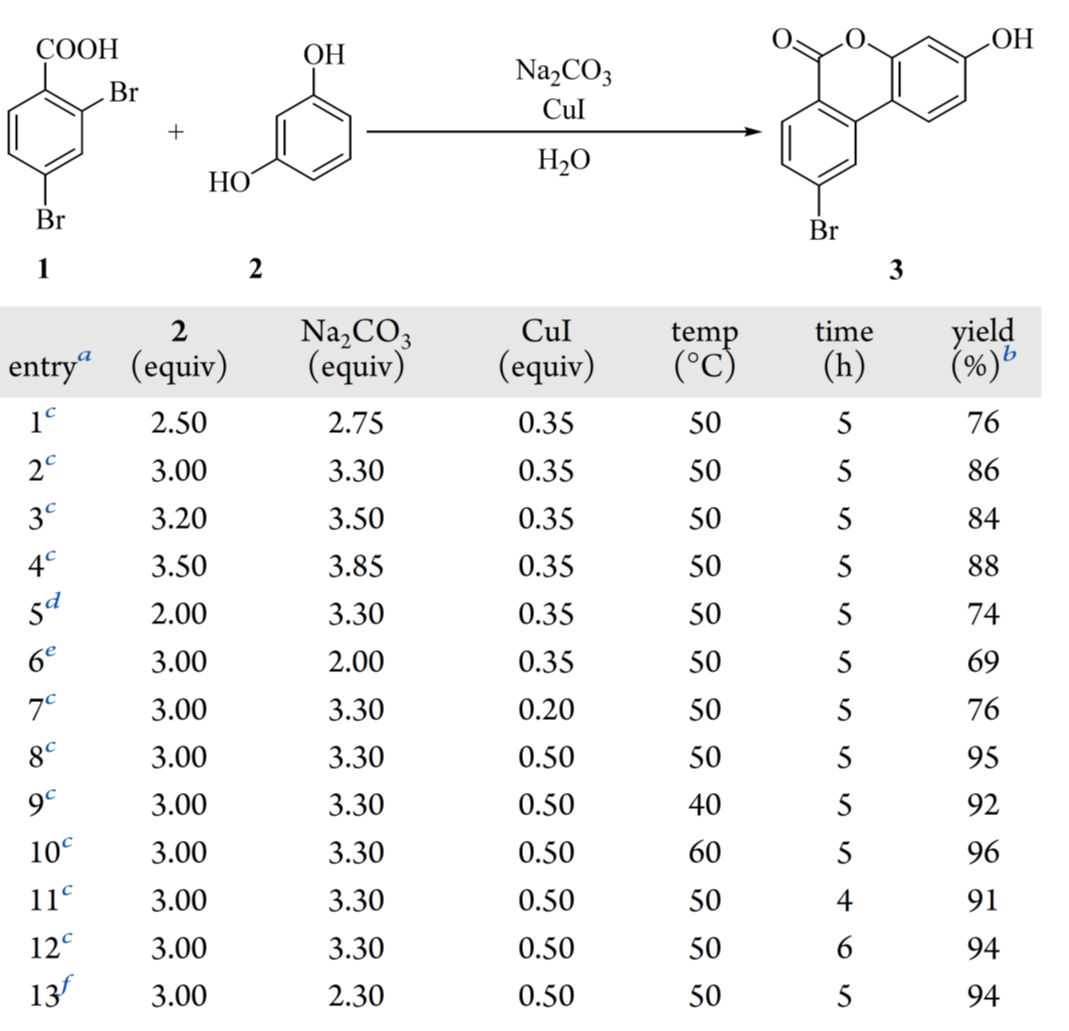
表2 Hurtley 反应条件筛选
6H-苯并[c]色烯-6-酮 3 虽可通过过滤和水洗初步分离,但残留铜确难以清除。文献报道其内酯环在碱性条件下可逆开环溶于碱液,酸化后闭环析出。据此,开发了改进的后处理工艺(图 4):反应结束后,向悬浮液中加入 4 M NaOH(8 V/W)和水(20 V/W),使有机组分(除铜杂质外)全部溶解,滤除不溶铜渣;滤液含开环钠盐 3′,加入 CH₃CN(4 V/W)以增溶未反应的间苯二酚(2),避免其在酸化时共沉淀;随后快速加入 12 M HCl(5 V/W)闭环,定量再生 3,直接过滤、水洗即得高纯产物。该工艺应用于 20.0 g 2,4-二溴苯甲酸(1)与 23.6 g 间苯二酚(2)的 Hurtley 反应,以 85% 收率获得 3,HPLC 纯度 99%,铜残留仅 4 ppm。
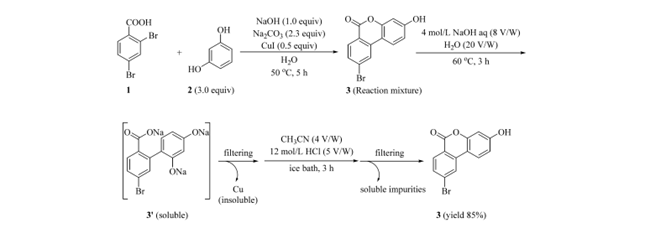
图4 Hurtley Reaction的后处理
2)6H-Benzo[c]chromen 4的工艺优化
鉴于成本低、适用性广,作者选用 BH₃·THF 作为替代还原剂。相比苯基硅烷/GaBr₃ 体系,BH₃·THF 可高选择性地将 6H-苯并[c]色烯-6-酮(3)还原为 6H-苯并[c]色烯(4),副产物极少。当 BH₃·THF 用量低于 3.00 eq.时,产率显著下降(表 3,entries 1–2);增至 4.00 或 5.00 eq.未提升产率(entries 2–4);升温至 40 或 60 °C 亦无改善(entries 5–6);缩短至 12 h 导致转化不足,延长反应时间则产率持平(entries 7–8)。故选定entry 2 条件用于放大。BH₃·THF 的淬灭是关键安全步骤:因 BH₃ 高度活泼,淬灭不彻底可能在后续萃取或减压浓缩中突发氢气释放,引发严重风险。标准操作为:冰浴冷却反应液至 0–5 °C,缓慢滴加甲醇至无气体逸出,再进行后续处理。粗品经萃取、浓缩后可直接用于下一步反应——实验证实,本步杂质不影响下游转化效率;而纯化(浆洗或重结晶)不仅收率低(重结晶仅 48%,柱层析 73%)、纯度未达优(重结晶 98%,但颜色差),反而增加操作风险与损耗。
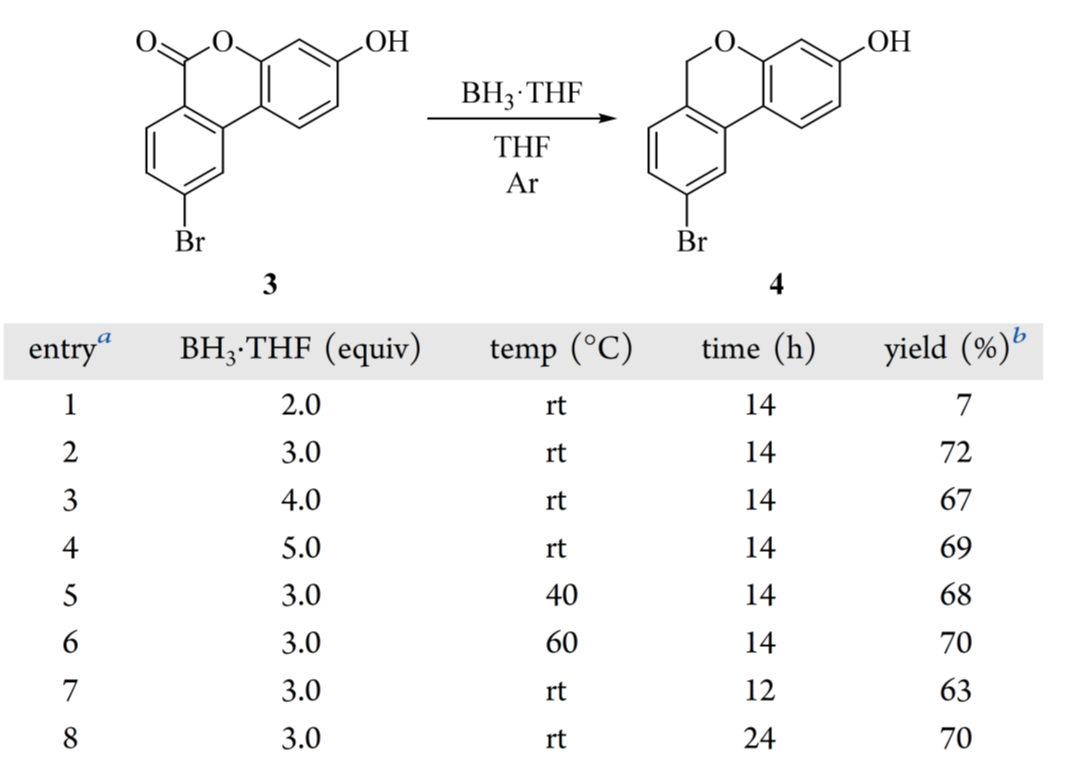
表3还原反应条件筛选
3)Propanamide 7的工艺优化
前人研究表明,(R)-1-氨基-1-氧代丙-2-基甲磺酸酯(10)与 TMG 在 CH₃CN 中可高效实现酚羟基的乳酰胺化,且消旋化程度极低。化合物 10 按“反应物合成”部分制备;随后在 CH₃CN 中考察其与 6H-苯并[c]色烯(4)的亲核取代反应(表 4)。筛选均相有机碱发现:NMM 和 DIPEA 因碱性不足,反应不发生(entries 1–2);TMG 给出 94% 收率和 97.4:2.6 e.r.,DBU 虽达 100% 收率,但 e.r. 降至 93.4:6.6(entries 3–4)。综合考量,TMG 为最优选择——兼顾高收率与优异对映体控制。 当量优化表明:10 eq. 低于 3.00 equiv(如 2.00 equiv)导致收率跌至 88%(entries 3、5–6);增至 3.00 equiv 无增益。TMG 用量减至 1.50 equiv 时收率亦为 88%,升至 2.50 equiv 则收率提升至 98%(entries 7–8);此时 e.r. 的轻微下降(<1%)可接受,因收率显著提高。
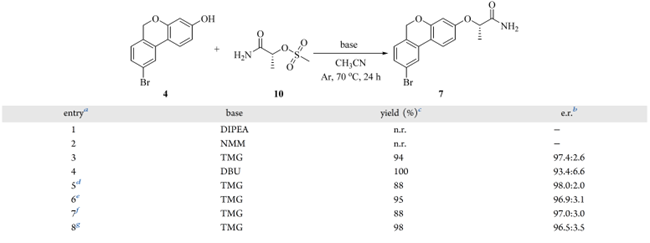
表4亲核取代反应条件筛选
丙酰胺(7)水溶性差,而其他杂质在碱性水相中保持溶解。反应结束后冷却至室温,加水稀释;减压共沸蒸除 CH₃CN,7 即析出;过滤、水洗、干燥,即得产物,收率 98%,纯度 98%(HPLC)。
经优化,亲核取代反应直接采用上步还原所得粗品 4 进行。结果证实:还原杂质不干扰取代反应,但影响丙酰胺(7)纯化——水沉淀法无法完全去除。改用 MTBE 溶剂重浆(reslurry),可高效脱除杂质。最终确立后处理为:水沉淀 → MTBE 重浆。
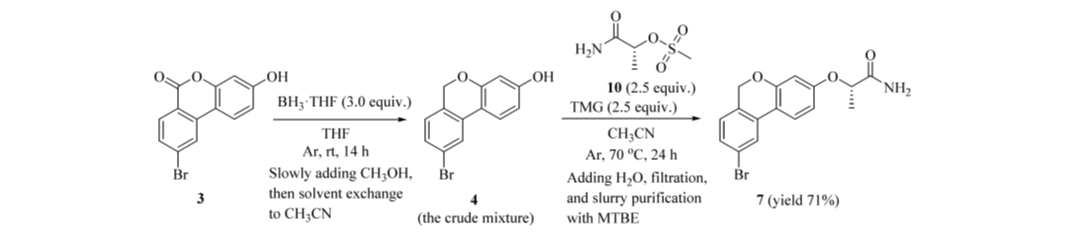
图5 两步一锅串联工艺
为规避中间体 6H-苯并[c]色烯(4)纯化收率低的问题,开发了从 6H-苯并[c]色烯-6-酮(3)至丙酰胺(7)的两步一锅串联工艺(图5):BH₃·THF 反应淬灭后,直接置换溶剂为 CH₃CN,不经分离,立即投入甲磺酸酯(10)进行取代。以 17.4 g 起始原料(3)计,丙酰胺(7)以 71% 总收率、98% HPLC 纯度(A%)、96.5:3.5 e.r. 直接获得,呈浅黄色粉末。相较原药物化学路线(3 → 4 → 7,三步,57% 总收率,含两次柱层析),该串联工艺减少一步操作,总收率提升 14 个百分点,并彻底避免柱层析。
5)Borate Ester 8的工艺优化
基于第一代路线,PdCl₂(dppf) 降至 2 mol% 后,收率反升至 95%(表 5,entry 1),证实其充足性。溶剂筛选(表 5,entries 2–6)显示:2-MeTHF 94%收率,与二氧六环相当;THF 因沸点偏低收率下降;EtOH、i-PrOH 和 EtOAc 则因副反应增多导致收率降低。最终确立最优条件:丙酰胺(7)、B₂Pin₂(1.50 equiv)、KOAc(2.00 equiv)、PdCl₂(dppf)(2 mol%)在 2-MeTHF 中,Ar 气保护下 80 °C 反应 6 h。反应结束后,混合物经硅藻土垫过滤除杂;滤液萃取、减压浓缩得粗品;粗品悬浮于正庚烷中重浆,脱除残留 B₂Pin₂;10 g 规模下直接获得硼酸酯(8),94%收率, 94%纯度(HPLC A%)。
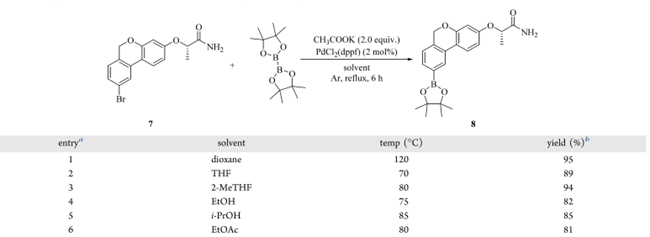
表5 Miyaura 硼化反应条件筛选
6)XJTU-L453的工艺优化
基于第一路线,在初始条件下(二氧六环/H₂O 5:1,3 mol% PdCl₂(dppf),1.5 equiv 嘧啶 9,2.0 equiv K₂CO₃)以89%的收率获得产物(表 6,entry 1)。其他钯催化剂(PdCl₂(dbtpf)、Pd(PPh₃)₄、Pd(dba)₂/2.4PCy₃)及碱(Na₂CO₃、K₃PO₄、Cs₂CO₃、t-BuONa)的筛选进一步确认:PdCl₂(dppf)/K₂CO₃ 仍为最优组合。
为规避高毒、难回收的二氧六环,作者评估了多种工业友好溶剂(表 6,entries 1–5):i-PrOH、EtOH、THF 和 2-MeTHF 收率均低于二氧六环,主因沸点偏低。i-PrOH/H₂O(5:1)表现相对最佳,但调整两相比例(entries 6–8)导致收率下降约 10%。提高 PdCl₂(dppf) 至 6 mol% 和 9 mol% 后,收率升至 76% 和 78%(entries 9–10);但钯残留风险同步上升。权衡后选定 i-PrOH/H₂O(5:1)中 6 mol% PdCl₂(dppf) 为折中条件。
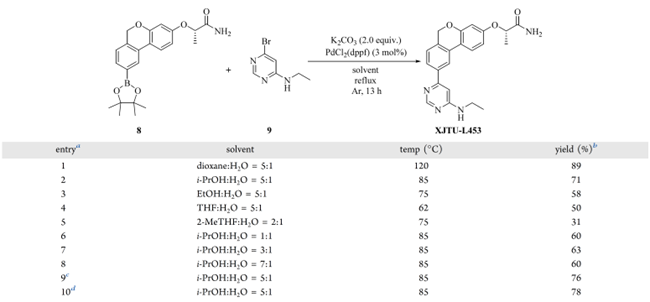
表6 Suzuki反应条件筛选
进一步延长反应时间无显著增益(表 7,entires 1–3)。碱筛选显示:K₃PO₄ 和 Na₂CO₃ 降低收率(entires 4–5),而 Cs₂CO₃(85%)和 t-BuONa(83%)提升收率(entires 6–7);其中 Cs₂CO₃ 效果最接近原二氧六环体系(89%)。最终优化条件为:嘧啶(9)(1.50 equiv)、Cs₂CO₃(2.00 equiv)、PdCl₂(dppf)(6 mol%)在 i-PrOH/H₂O(5:1)中,Ar 气保护下 85 °C 反应 13 h。
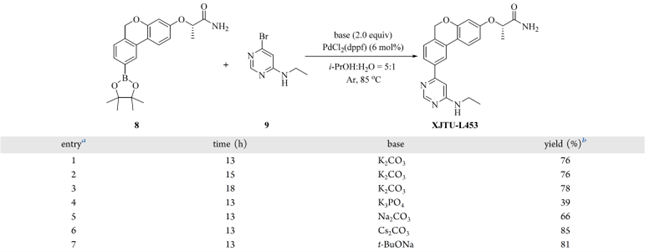
表7 Suzuki反应条件筛选
同时,对残留钯的去除也做了研究。即评估了三种含硫清除剂:N-乙酰半胱氨酸、L-半胱氨酸和 Silia-Thiol。三者均通过–SH 基团与钯形成稳定 Pd–S 络合物实现捕获。其中 N-乙酰半胱氨酸效率最高,L-半胱氨酸次之;Silia-Thiol 单独使用效果差(entries 2–4)。采用“N-乙酰半胱氨酸预处理 + Silia-Thiol”两步法,钯分别降至 41 ppm 和 56 ppm(entries 5–6),仍高于 10 ppm 限值;对前者再追加一次 Silia-Thiol 处理,钯降至 9 ppm,达标entry 7)。最终确立的钯清除工艺如下:粗反应液经硅藻土过滤、EtOAc 萃取、减压浓缩;残渣溶于 2-MeTHF,加入 N-乙酰半胱氨酸、K₃PO₄ 和 H₂O,60 °C 搅拌 5 h 后分液;有机相换溶剂为 i-PrOH(兼顾 XJTU-L453 溶解性与 Silia-Thiol 分散性),加入 Silia-Thiol,60 °C 再搅拌 5 h,过滤;重复该吸附步骤一次,5 g 规模下残留钯 <10 ppm。
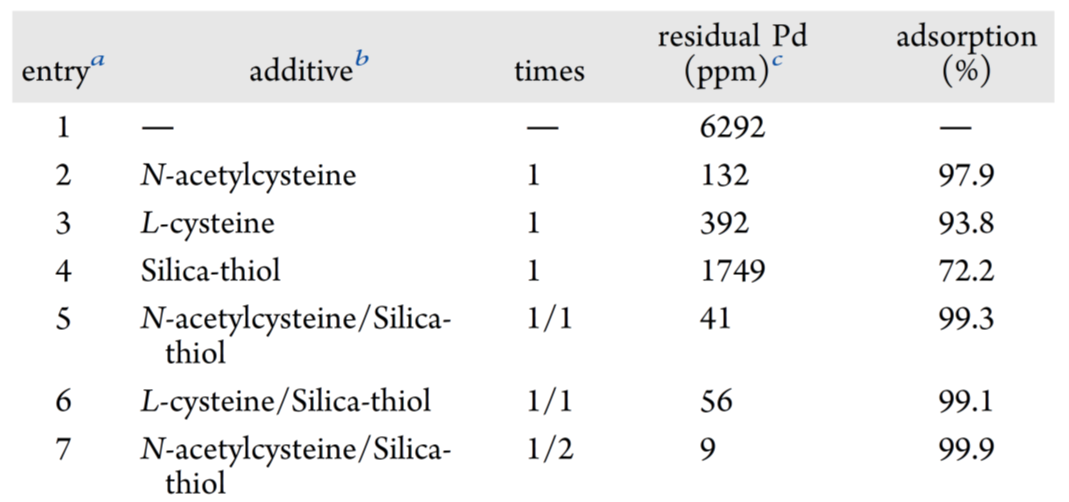
表8 残留钯的去除实验
7)XJTU-L453结晶工艺
钯去除后,浓缩物经 MeOH/DCM(5:1)研磨纯化,得灰白色 XJTU-L453,收率 85%,纯度 92%(HPLC A%)。二次研磨仅小幅提升纯度,故转向结晶工艺开发。基于室温 HPLC 溶解度测定(图6),筛选溶解度 5.0–20.0 mg/mL 的溶剂:n-BuOH(5 V)、2-MeTHF(12 V)几乎不析晶(表 9,entires 1、3);i-PrOH(8 V)得浅黄色晶体,回收率 61%,纯度 96%(entry 2)。 进一步考察溶解度 2.0–4.0 mg/mL 的溶剂,仅 EtOH(40 V)获得白色晶体,回收率 70%,纯度 99%(entry10)。
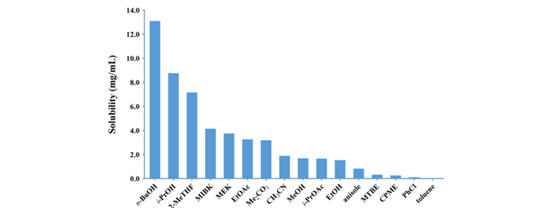
图6 XJTU-L453溶解性实验
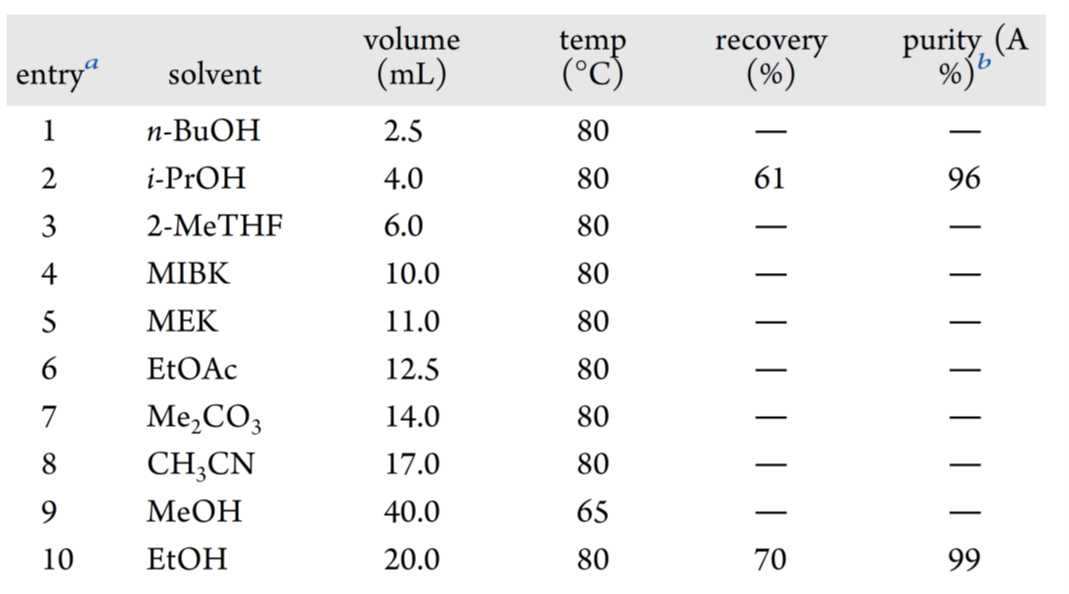
表9 XJTU-L453的结晶实验
三批粗品(各 5.00 g)在 200 mL EtOH 中结晶,回收率稳定在 68–71%;所得白色固体纯度 99%,含两种杂质(0.15% 和 0.84%,HPLC A%),e.r. 99.4:0.6,Pd <10 ppm(图 7)。铃木偶联–钯清除–EtOH 重结晶集成工艺,在 10 g 规模下总收率 60%,获得高纯度 XJTU-L453。

图7 XJTU-L453晶型
参考资料
Efficient and Scalable Synthesis of a Novel Selective PI3Kα Inhibitor XJTU-L453
Peng Xiang, Yanhong Chen, Xinyuan Zhang, Zitian Li, Songyao Wang, Yiping Li*
Org. Process Res. Dev. 2026. DOI: 10.1021/acs.oprd.5c00415
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
关注Chem-Station抖音号:79473891841
请登陆TCI试剂官网查看更多内容
https://www.tcichemicals.com/CN/zh/




















No comments yet.