
本文作者:杉杉
导读
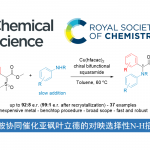
实用的C-H/C-H偶联反应,作为一个具有挑战性且具吸引力的合成技术,可避免两个偶联底物的预功能化,同时生成所需的C-C键。近日,美国斯克利普斯研究所余金权教授课题组在J. Am. Chem. Soc.发表论文,报道了使用基于环戊烷的N-保护的β-氨基酸配体,实现游离脂肪酸的C(sp3)-H/C(sp2)-H偶联反应。该反应使用廉价的过碳酸钠(Na2CO3·1.5H2O2)作为唯一氧化剂,并生成水作为唯一的副产物。通过此方案可以轻松合成一系列生物学上重要的骨架,如四氢化萘(tetralins),苯并二氢吡喃(chromanes)和茚满(indanes)。最后,通过四步简明(±)-russujaponol F全合成,进一步证明了反应的实用性。
Rapid Construction of Tetralin, Chromane, and Indane Motifs via Cyclative C-H/C-H Coupling: Four-Step Total Synthesis of (±)-Russujaponol F
Zhe Zhuang, Alastair N. Herron, Shuang Liu, and Jin-Quan Yu*
JAm. Chem. Soc.ASAP DOI:10.1021/jacs.0c12484
正文
碳-碳键的形成作为有机合成中最重要的反应类型之一。由于具有缩短合成的潜力,在过去二十年中,使用C-H活化构建C-C键的策略迅速发展。虽然大多数偶联方法都需要预先官能化的偶联底物(如有机硼和有机卤化物),但C-H/C-H偶联反应提供了一种补充策略,可直接从两个简单的C-H键中构建C-C键(Scheme 1A)。与传统的偶联方法相比,这种绿色且原子经济的方法极更具吸引力,因为水可能是该过程唯一的化学计量副产物。迄今为止,大量的研究主要集中在两个相对更活泼的C(sp2)-H键上,用于联芳基的合成,仅有少数反应用于构建更具挑战性的C(sp3)-C(sp2)键。由于此类反应需要导向基团来促进环金属化(cyclometalation),因此需额外的步骤来引入和除去导向基团。同时,此类方法也存在一定的局限性,如化学计量使用贵金属银盐和苛刻的条件。此外,由C(sp3)-H活化引发的C(sp3)-H/C(sp2)-H偶联很大程度上限于更具反应性的杂环C(sp2)-H键。尽管C-H/C-H偶联反应对于有机合成具有巨大的价值,但同时使用实用的氧化剂和天然底物的C(sp3)-H/C(sp2)-H偶联反应,仍具有巨大的挑战。
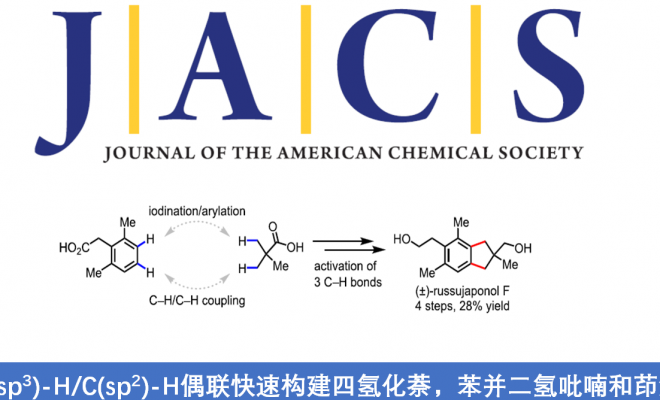
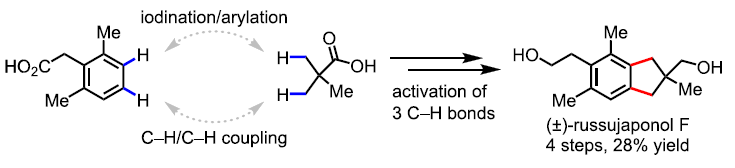
对于提高C-H功能化策略的合成效率,需满足三个条件:(1)能够使用简单的底物来合成多种天然产物能力;(2)使用固有的基团作为导向基团;(3)精确控制C-H官能团化的位点选择性。然而,满足这些条件的方法难以执行,仅有少数文献报道[1]。为了解决这些挑战,余金权教授课题组报道了一种使用固有的游离羧酸作为导向基团的C(sp3)-H/C(sp2)-H偶联反应(Scheme 1B)。在该反应中,使用环戊烷基的N-保护的β-氨基酸配体和实用且廉价的氧化剂(过碳酸钠)。同时,可合成一系列天然产物中常见的骨架,如四氢化萘、苯并二氢吡喃和茚满(Figure 1)。此外,作者以苯乙酸和新戊酸为底物,通过四步简明的(±)-russujaponol F全合成(迄今报道的最短和最高收率的路线)[2],进一步证明了反应的实用性。
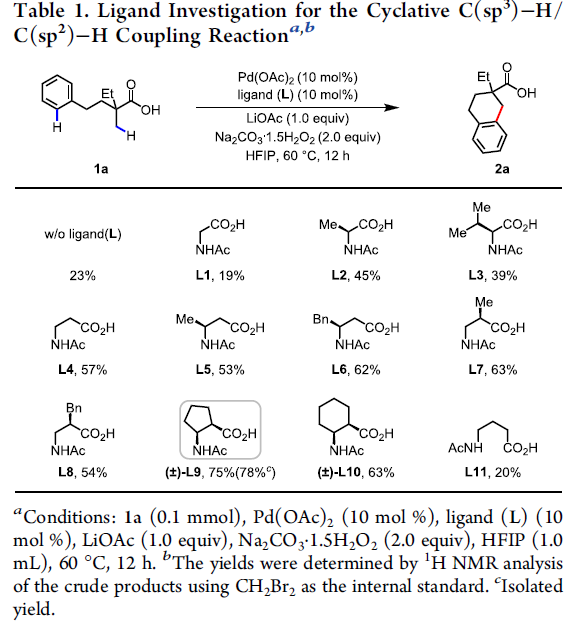
首先,作者以脂族酸1a作为模型底物,对C(sp3)-H/C(sp2)-H偶联反应进行了条件筛选(Table 1)。反应的最佳条件为:以Pd(OAc)2为催化剂,(±)-L9为配体, LiOAc为金属添加剂,过碳酸钠为氧化剂,六氟异丙醇为溶剂时,在60 ℃下反应,即可获得78%收率的目标产物2a。
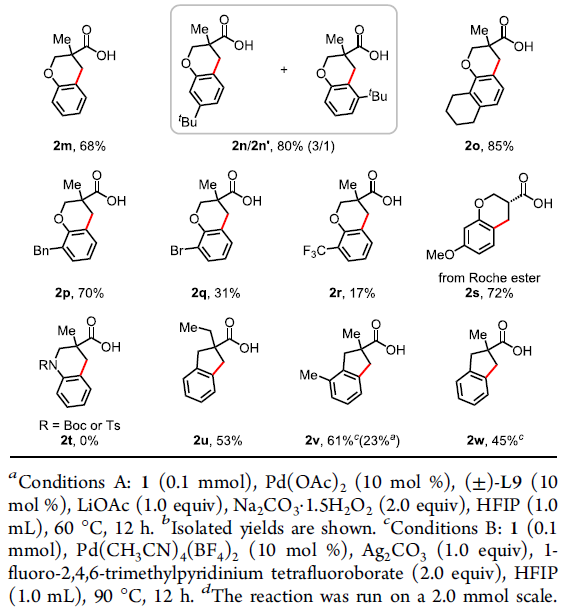
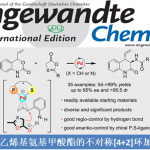
在获得上述最佳反应条件后,作者开始对底物1进行了扩展(Table 2)。各种带有单个α-甲基(1a–1e和1h)或α-偕-二甲基(1f和1g)的叔脂肪酸都可以作为反应物,以中等到良好的收率获得四氢化萘产物。反应性较低的含有α-氢的游离羧酸,也以35-63%的收率合成产物2i–2l。其中,芳环上可耐受各种官能团,如甲基、甲氧基、氟、氯、萘基等。其次,该方法也可扩展到苯并二氢吡喃的合成,如2m–2s。芳环(2s和2n–2p)上带有一系列供电子基团(甲氧基、叔丁基、环己基和苄基)时,均以良好的收率(70-85%)获得所需的产物,而含有吸电子基团(溴和三氟甲基)(2q和2r)显示出较低的反应性(31%和17%),这可能是由于缺电子的芳烃的C(sp2)-H活泼性不高所致。值得注意的是,对于含有间甲氧基的脂肪酸(1j和1s),具有很高的区域选择性,而带有叔丁基(1n)的底物提供了区域异构体的混合物(2n/2n’=3/1)。然而,对于N-Boc或N-Ts保护的羧酸1t无法获得四氢异喹啉产物2t。此外,该偶联反应也适合于茚满骨架的合成,如2u–2w。
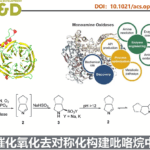
Illudalane倍半萜烯构成一大类天然产物,通常带有具有挑战性的全碳四元中心的茚满骨架(Scheme 2A)。由于它们具有潜在的生物学活性,研究者已开发出多种合成方法[3-4]。鉴于此方法可用于构建茚满骨架,作者着手通过多个C-H功能化来进行(±)-russujaponol F的全合成(Scheme 2B)。Baudoin课题组[5]报道了基于C(sp3)-H芳构化策略,实现外消旋和对映选择性形式的russujaponol F的首次全合成,分13步(收率26%)和15步(收率12%)。作者以市售的苯乙酸3为初始原料,经酯化和碘化后,获得79%收率的芳基碘化物4。随后,使用Pd(OAc)2和配体L12,获得62%收率的单芳基化产物5,同时也形成12%收率的偶联产物6。若使用1-氟-2,4,6-三甲基吡啶四氟化硼作为氧化剂,也可将5转化为偶联产物6,收率为41%。最后,经LAH还原,即可获得96%收率的(±)-russujaponol F。总的来说,该反应只需四步,即可获得28%收率的(±)-russujaponol F,作为迄今为止最短且收率最高的合成路线。

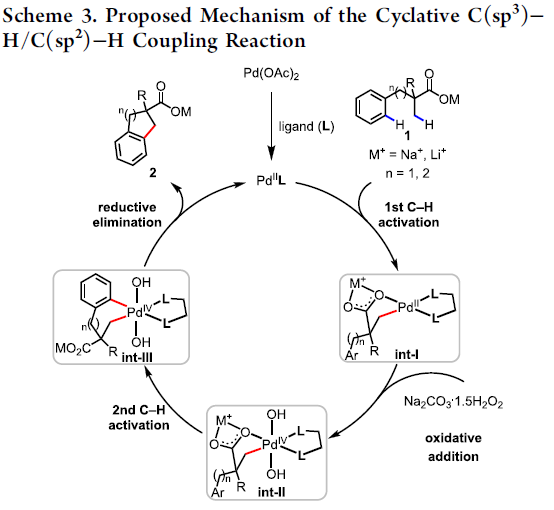
根据相关文献的查阅[5-7]以及对游离酸C-H活化反应的研究[3],作者提出了一种可能的C(sp3)-H/C(sp2)-H偶联反应机理(Scheme 3)。首先,Pd(OAc)2与MPAA配体的配位,产生了活性LPd(II)。酸性底物1与Pd配位后,Na+或Li+抗衡阳离子和MPAA配体均会加速β-C(sp3)-H键的环钯化形成int-I。紧接着,int-I经氧化加成生成int-II。随后,int-II侧链上的苯基经历二次C(sp2)-H活化,形成七元或六元的钯环中间体int-III,同时,也可能在int-I生成int-III的氧化加成之前进行二次C-H活化。最后,int-III经还原消除得到偶联产物2,并再生了LPd(II)配合物。
总结
余金权教授课题组报道了一种Pd(II)催化的C(sp3)-H/C(sp2)-H偶联反应。其中,该反应以环戊烷基N-保护的β-氨基酸为配体,以廉价的过碳酸钠作为唯一氧化剂,以固有的游离羧酸作为导向基团。同时,该方案可合成一系列生物学上重要的骨架,如四氢化萘、苯并二氢吡喃和茚满。此外,通过简单的四步(±)-russujaponol F全合成,进一步证明了该反应的实用性。
参考文献
[1] For selected examples of total synthesis using multiple C-H functionalizations, see: (a) Wang, D.-H.; Yu, J.-Q. Highly convergent total synthesis of (+)-lithospermic acid via a late-stage intermolecular C-H olefination. J. Am. Chem. Soc. 2011, 133, 5767−5769. (b) Gutekunst, W. R.; Baran, P. S. Total synthesis and structural revision of the piperarborenines via sequential cyclobutane C-H arylation. J. Am. Chem. Soc. 2011, 133, 19076−19079. (c) Rosen, B. R.; Simke, L. R.; Thuy-Boun, P. S.; Dixon, D. D.; Yu, J.-Q.; Baran, P. S. C-H functionalization logic enables synthesis of (+)-hongoquercin A and related compounds. Angew. Chem., Int. Ed. 2013, 52, 7317−- (d) Hong, B.; Li, C.; Wang, Z.; Chen, J.; Li, H.; Lei, X.Enantioselective total synthesis of (−)-incarviatone A. J. Am. Chem.Soc. 2015, 137, 11946−11949. (e) Dailler, D.; Danoun, G.; Ourri, B.; Baudoin, O. Divergent synthesis of aeruginosins based on a C(sp3)−H activation strategy.Chem. – Eur. J. 2015, 21, 9370−9379. (f) Wu,
F.; Zhang, J.; Song, F.; Wang, S.; Guo, H.; Wei, Q.; Dai, H.; Chen, X.; Xia, X.; Liu, X.; Zhang, L.; Yu, J.-Q.; Lei, X. Chrysomycin A derivatives for the treatment of multi-drug-resistant tuberculosis. ACS Cent.Sci. 2020, 6, 928−938.
[2] Gaich, T.; Baran, P. S. Aiming for the ideal synthesis. J. Org. Chem. 2010, 75, 4657−4673. [3] (a) Melot, R.; Craveiro, M.; Bürgi, T.; Baudoin, O. Divergent enantioselective synthesis of (nor)illudalanesesquiterpenes via Pd0-catalyzed asymmetric C(sp3)−H activation. Org. Lett. 2019, 21, 812−815. (b) Melot, R.; Craveiro, M. V.; Baudoin, O. Total synthesis of (nor)illudalanesesquiterpenes based on a C(sp3)−H activation strategy. J. Org. Chem. 2019, 84, 12933−12945. [4] For recent examples, see: (a) Tiong, E. A.; Rivalti, D.; Williams, B. M.; Gleason, J. L. A concise total synthesis of (R)-puraquinonic acid. Angew.Chem., Int. Ed. 2013, 52, 3442−3445. (b) Elmehriki, A. A. H.; Gleason, J. L. A spiroalkylation method for the stereoselective construction of α-quaternary carbons and its application to the total synthesis of (R)-puraquinonic acid. Org. Lett. 2019, 21, 9729−9733. (c) Zeng, Z.; Zhao, Y.; Zhang, Y. Divergent total syntheses of five illudalanesesquiterpenes and assignment of the absolute configuration. Chem. Commun. 2019, 55, 4250−4253. (d) Xun, M. M.; Bai, Y.; Wang, Y.; Hu, Z.; Fu, K.; Ma, W.; Yuan, C. Synthesis of four illudalanesesquiterpenes utilizing a one-pot Diels−Alder/oxidative aromatization sequence. Org. Lett. 2019, 21, 6879−6883. [5] For early examples of C(sp2)−H/C(sp2)−H coupling reactions, see: (a) Stuart, D. R.; Fagnou, K. The catalytic cross-coupling of unactivatedarenes. Science 2007, 316, 1172−1175. (b) Xia, J.-B.; You, S.-L. Carbon−carbon bond formation through double sp2 C-H activations: synthesis of ferrocenyloxazoline derivatives. Organometallics2007, 26, 4869−4871. (c) Hull, K. L.; Sanford, M. S. Catalytic and highly regioselective cross-coupling of aromatic C-H substrates. J. Am. Chem. Soc. 2007, 129, 11904−11905. (d) Brasche, G.; García-Fortanet, J.; Buchwald, S. L. Twofold C-H functionalization: palladium-catalyzedorthoarylation of anilides. Org. Lett. 2008, 10, 2207−2210. (e) Cho, S. H.; Hwang, S. J.; Chang, S. Palladiumcatalyzed C-H functionalization of pyridine N-oxides: highly selective alkenylation and direct arylation with unactivatedarenes. J. Am. Chem. Soc. 2008, 130, 9254−9256. (f) Zhao, X.; Yeung, C. S.; Dong, V. M. Palladium-catalyzed ortho-arylation of O-phenylcarbamates with simplearenes and sodium persulfate. J. Am. Chem. Soc. 2010, 132, 5837−5844. (g) Wang, X.; Leow, D.; Yu, J.-Q. Pd(II)-catalyzed paraselective C-H arylation of monosubstitutedarenes. J. Am. Chem. Soc. 2011, 133, 13864−13867.
[6] For Pd-catalyzed C(sp3)−H/C(sp2)−H coupling reactions initiated by C(sp2)−H activation, see: (a) Liégault, B.; Fagnou, K. Palladium-catalyzed intramolecular coupling of arenes and unactivated alkanes in air. Organometallics 2008, 27, 4841−4843. (b) Pierre, C.; Baudoin, O. Intramolecular PdII-catalyzed dehydrogenative C(sp3)−C(sp2) coupling: an alternative to Pd0-catalyzed C(sp3)−H arylation from aryl halides? Tetrahedron 2013, 69, 4473−4478. (c) Shi, J.-L.; Wang, D.; Zhang, X.-S.; Li, X.-L.; Chen, Y.-Q.; Li, Y.-X.; Shi, Z.-J. Oxidative coupling of sp2 and sp3 carbon−hydrogen bonds to construct dihydrobenzofurans. Nat. Commun. 2017, 8, 238. [7] For Pd-catalyzed C(sp3)−H/C(sp2)−H coupling reactions initiated by C(sp3)−H activation, see: (a) Jiang, Y.; Deng, G.; Zhang, S.; Loh, T.-P. Directing group participated benzylic C(sp3)−H/C(sp2)−H cross-dehydrogenative coupling (CDC): synthesis of azapolycycles. Org. Lett. 2018, 20, 652−655. (b) Sun, W.-W.; Liu, J.-K.; Wu, B. Practical synthesis of polysubstitutedunsymmetric 1,10-phenanthrolines by palladium catalyzed intramolecular oxidative cross coupling of C(sp2)−H and C(sp3)−H bonds of carboxamides. Org. Chem. Front. 2019, 6, 544−550. (c) Hao, H.-Y.; Mao, Y.-J.; Xu, Z.-Y.; Lou, S.-J.; Xu, D.-Q. Selective cross-dehydrogenativeC(sp3)−H arylation with arenes. Org. Lett. 2020, 22, 2396−2402.



















No comments yet.