
本文作者:杉杉
导读:
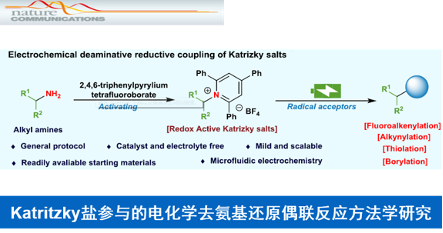
南京大学的王毅等研究团队与南京工业大学的郭凯团队共同报道一种全新的通过Katritzky盐参与的电化学去氨基官能团化策略,进而成功应用于一系列后续的氟烯基化 (fluoroalkenylation)、炔基化以及硫醚化 (thiolation)反应方法学的相关研究。这一全新的电化学去氨基官能团化策略中,无需加入催化剂以及相应的支持电解质。同时,具有反应条件温和、优良的SET化学应用价值以及良好的官能团兼容性等优势。
Electrochemical C-N bond activation for deaminative reductive coupling of Katritzky salts
Liu, X. Tao, Y. Mao, X. Yuan, J. Qiu, L. Kong, S. Ni, K. Guo, Y. Wang, Y. Pan, Nat. Commun. 2021, 12, 6745. doi: 10.1038/s41467-021-27060-7.

正文
近年来,有机电化学合成的相关研究,已经备受诸多研究团队的广泛关注[1]。这里,受到通过采用牺牲廉价易得的金属阳极促进的SET还原过程,形成相应自由基中间体的相关研究[2]-[5]以及前期本课题组对于采用电化学反应条件促进的通过氧化还原活性酯参与的脱羧还原偶联反应方法学 (Fig. 1a)[2]、通过三级醇衍生的烷基肼基甲酸酯 (tertiary alcohol-derived alkyl carbazate)参与的电化学去氧官能团化反应 (deoxygenative functionalization, Fig. 1b)[6]、通过一系列Katritzky盐参与的有机合成转化[7]以及将微流体技术 (microfluidic technique)应用于SET氧化还原中性有机电合成化学[8]相关研究报道的启发,南京大学的王毅等研究团队与南京工业大学的郭凯团队共同设计出一种全新的通过Katritzky盐参与的电化学去氨基还原偶联策略,并进一步将上述策略应用于一系列氟烯基化、炔基化以及硫醚化反应方法学的相关研究 (Fig. 1c)。

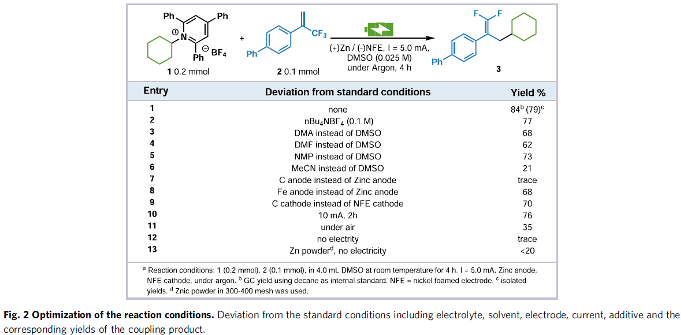
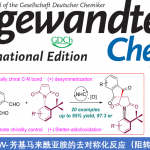
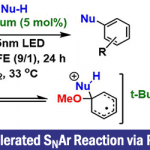
首先,作者采用Katritzky盐1a与α-三氟甲基烯基化合物2作为模型底物,进行氟烯基化反应条件的优化筛选(Fig. 2)。进而确定最佳的氟烯基化反应条件为:采用Zn作为阳极,NFE (nickel foam electrode)作为阴极,控制电流为5 mA,DMSO作为反应溶剂,反应温度为室温,最终获得84%收率的氟烯基化产物3。
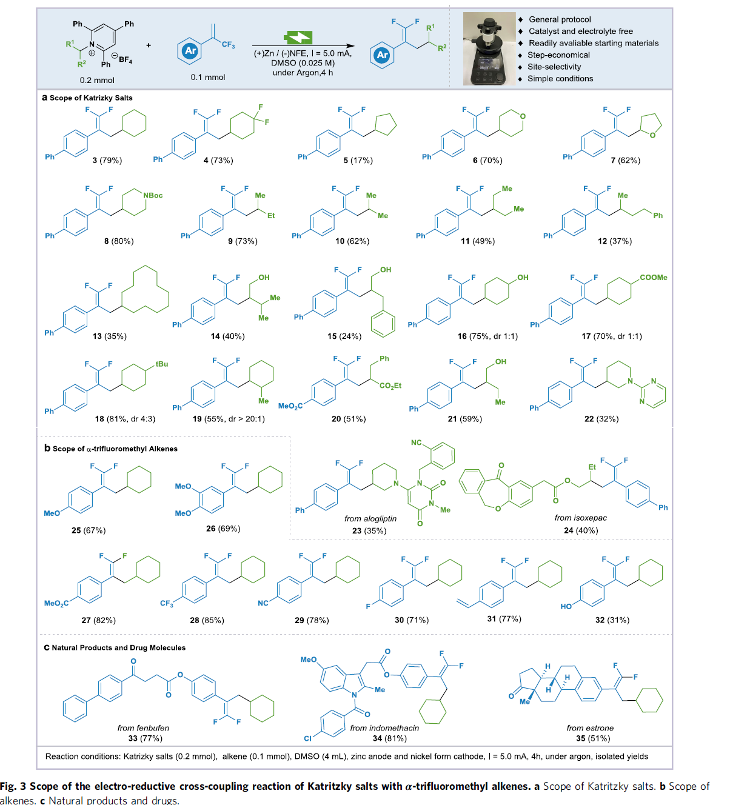
在上述的最佳反应条件下,作者首先对各类Katritzky盐底物的应用范围进行考察 (Fig. 3)。研究表明,一系列一级与二级Katritzky盐均能够较好地与上述的氟烯基化反应条件兼容,并以中等至良好的反应收率,获得相应的目标产物3–24 (Fig. 3a)。之后,该小组进一步对一系列α-三氟甲基烯基底物 (作为自由基受体)的应用范围进行深入研究。作者发现,上述的氟烯基化反应条件对于一系列芳基中具有供电子与吸电子基团取代的α-三氟甲基烯基底物以及部分生物活性分子的α-三氟甲基烯基衍生物,均能够有效地兼容,并以中等至良好的反应收率,获得相应的目标产物25–35,并表现出良好的官能团兼容性 (Fig. 3b-3c)。
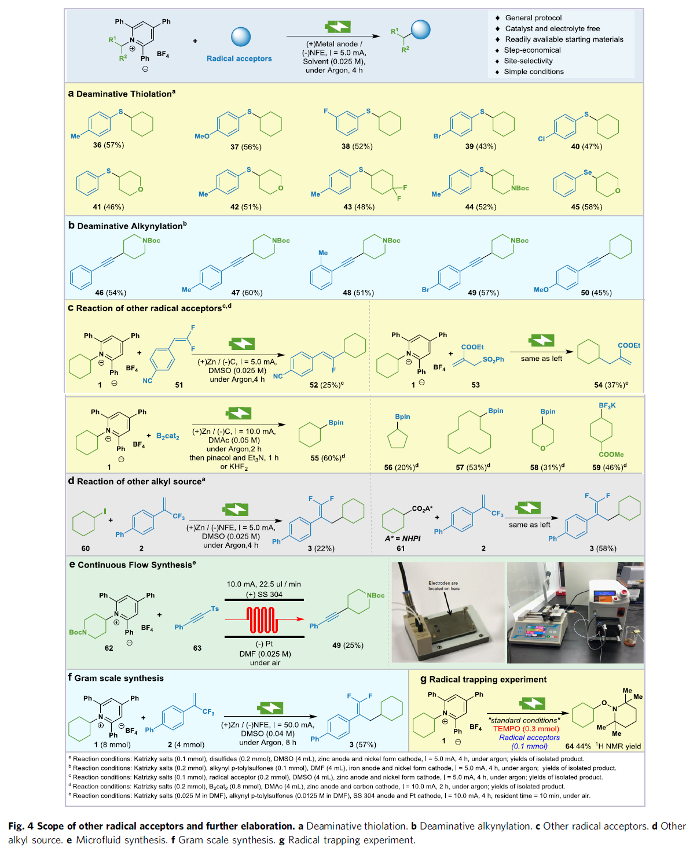
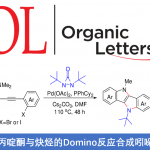
之后,作者进一步发现,这一全新的电化学去氨基官能团化策略同样能够有效地应用于一系列硫醚化 (Fig. 4a)、硒醚化 (Fig. 4a)、炔基化 (Fig. 4b)以及其他不同类型的合成转化方法学的相关研究 (Fig. 4c-4d)。同时,作者采用微流体技术电化学平台 (microfluidic electrochemistry platform)对上述的电化学去氨基官能团化方法学中的反应条件进行进一步优化,进而使上述合成转化过程中的可重现性与合成应用价值获得极大提升 (Fig. 4b)。之后,作者进一步通过克级规模实验的相关研究进一步表明,这一全新的电化学去氨基官能团化策略具有良好的合成应用价值 (Fig. 4f)。最终,作者通过自由基捕获实验的相关研究初步表明,上述的电化学去氨基官能团化过程中涉及自由基中间体的参与 (Fig. 4g)。
总结
南京大学的王毅等研究团队与南京工业大学的郭凯团队共同合作设计出一种全新的通过Katritzky盐参与的电化学去氨基还原偶联反应策略,并进一步应用于后续的氟烯基化 (fluoroalkenylation)、炔基化以及硫醚化(thiolation)反应方法学的相关研究。这一全新的电化学去氨基官能团化策略中,无需加入催化剂以及相应的支持电解质,同时,具有温和的反应条件、高度的官能团兼容性以及良好的合成应用价值等优势。
参考文献
- [1] (a) M. Yan, Y. Kawamata, P. S. Baran, Chem. Rev. 2017, 117, 13230. doi: 10.1021/acs.chemrev.7b00397.
- (b) H. Wang, X. Gao, Z. Lv, T. Abdelilah, A. Lei, Chem. Rev. 2019, 119, 6769. doi: 10.1021/acs.chemrev.9b00045.
- [2] T. Koyanagi, A. Herath, A. Chong, M. Ratnikov, A. Valiere, J. Chang, V. Molteni, J. Loren, Org. Lett. 2019, 21, 816. doi: 10.1021/acs.orglett.8b04090.
- [3] K. Jiao, D. Liu, H. Ma, H. Qiu, P. Fang, T. Mei, Angew. Chem. Int. Ed. 2020, 59, 6520. doi: 10.1002/anie.201912753.
- [4] G. Kumar, A. Peshkov, M. Brzozowska, P. Nikolaienko, C. Zhu, M. Rueping, Angew. Chem. Int. Ed. 2020, 59, 6513. doi: 10.1002/anie.201915418.
- [5] R. C. Samanta, J. Struwe, L. Ackermann, Angew. Chem. Int. Ed. 2020, 59, 14154. doi: 10.1002/anie.202004958.
- [6] Y. Gao, Z. Wu, L. Yu, Y. Wang, Y. Pan, Angew. Chem. Int. Ed. 2020, 59, 10859. doi: 10.1002/anie.202001571.
- [7] (a) S. Plunkett, C. H. Basch, S. O. Santana, M. P. Watson, J. Am. Chem. Soc. 2019, 141, 2257. doi: 10.1021/jacs.9b00111.
- (b) F. J. R. Klauck, M. J. James, F. Glorius, Angew. Chem. Int. Ed. 2017, 56, 12336. doi: 10.1002/anie.201706896.
- (c) S. Ni, C. Li, Y. Mao, J. Han, Y. Wang, H. Yan, Y. Pan, Sci. Adv. 2019, 5, eaaw9516. doi: 10.1126/sciadv.aaw9516.
- (d) J. Liao, C. H. Basch, M. E. Hoerrner, M. R. Talley, B. P. Boscoe, J. W. Tucker, M. R. Garnsey, M. P. Watson, Org. Lett. 2019, 21, 2941. doi: 10.1021/acs.orglett.9b01014.
- [8] Y. Mo, Z. Lu, G. Rughoobur, P. Patil, N. Gershenfeld, A. I. Akinwande, S. L. Buchwald, K F. Jensen, Science 2020, 368, 1352. doi: 10.1126/science.aba3823.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载













No comments yet.