
本文作者:Joy
导读
扬州大学的段伟良教授报道首例通过全新的非对称双膦 (unsymmetric bisphosphine, PCP’)钳型镍配合物 (pincer-nickel complex)促进的一级膦与缺电子烯基化合物之间的不对称加成反应方法学,进而以高度的对映与非对映选择性 (99% ee, >20:1 dr)以及良好至优良的反应收率 (57-92%),成功完成一系列具有P–立体生成中心的二级膦-硼烷分子的构建。之后,该小组进一步将相应的二级膦-硼烷产物应用于通过卤代烷试剂参与的烷基化过程,进而获得各类具有多种不同官能团的P-手性有机分子。
Asymmetric Synthesis of P-Stereogenic Secondary Phosphine-Boranes by an Unsymmetric Bisphosphine Pincer-Nickel Complex
C. Wang, K. Huang, J. Ye, W. Duan, J. Am. Chem. Soc. 2021, 143, 5685. doi: 10.1021/jacs.1c02772.

正文
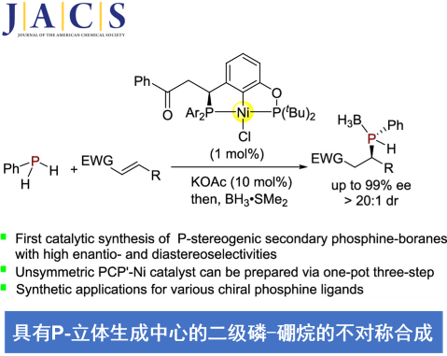
具有P-立体生成中心的有机分子,例如CAMP、DIPAMP、Tangphos、QuinoxPhos以及BIBPO等,作为重要的手性配体或有机催化剂,目前已经广泛应用一系列不同类型的不对称合成转化方法学的相关研究[1]-[4]。同时,有机合成化学家开始致力于发展一系列更为有效地策略,进而实现具有P-立体生成中心的三级膦分子的构建[5]-[10]。然而,与构型稳定的P–手性三级膦分子相比,P–手性二级膦分子则由于其较低的翻转能垒,能够较为容易地进行P–手性中心的外消旋化过程[11],进而使具有光学活性二级膦分子的合成面临诸多的挑战[12]-[13]。同时,研究发现,具有P–立体生成中心的二级膦化合物作为关键砌块,能够有效地应用于一系列手性磷配体[12]-[15]以及各类过渡金属配合物的构建[16] (Figure 1a)。而且,通过相应的催化不对称策略,直接构建具有P–生成中心的二级膦分子,目前尚未有相关的文献报道。为解决手性二级膦分子合成中存在的巨大挑战,本文中,作者首次报道通过全新的双膦 (unsymmetric bisphosphine, PCP’)钳型镍配合物 (pincer-nickel complex)参与的不对称加成反应方法学,进而顺利完成一系列具有P–立体生成中心的二级膦-硼烷分子的构建,并获得高度的对映与非对映选择性 (Figure 1b)。同时,作者发现,相应的P–手性二级膦-硼烷产物能够十分容易地转化为一系列不对称转化方法学研究中极为重要的手性膦配体。

首先,作者选择苯基膦与查尔酮作为模型底物,进行相关反应条件的优化筛选。进而确定最佳的反应条件为:采用(S)-4作为催化剂,乙酸钾作为碱,DCM作为反应溶剂,反应温度为-40 °C,并获得相应的膦氢化 (hydrophosphination)产物。之后,通过Me2S•BH3进行处理,最终以高度的对映以及非对映选择性 (94% ee, 13:1 dr)与优良的反应收率 (82%)获得具有P–立体生成中心的二级膦-硼烷产物。

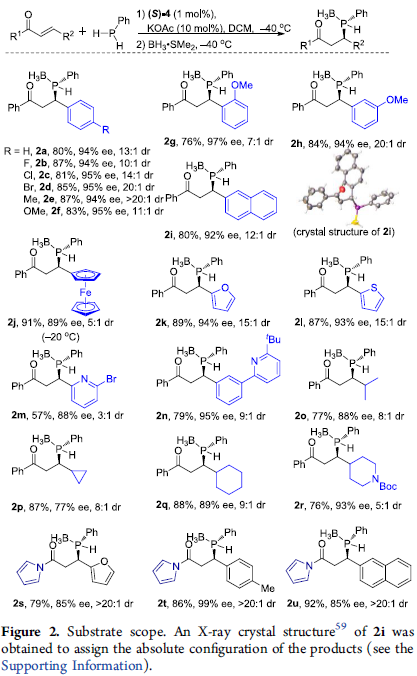
在上述的最佳反应条件下,作者开始对一系列烯酮底物的应用范围进行考察 (Figure 2)。研究发现,上述的标准反应条件,对于一系列芳基中具有吸电子与供电子基团取代的β-芳基-α,β-苯基酮底物,均能够良好地兼容,并以优良的对映与非对映选择性,获得相应的P–手性二级膦-硼烷产物 (2b-2f)。之后,该小组发现,上述的最佳反应条件对于β-芳基邻位或间位中的不同取代基团,同样能够有效地兼容,并获得相应的P–手性二级膦-硼烷产物2g与2h (97%反应收率,97% ee, >20:1 dr)。同时,作者发现,β-萘基、β-二茂铁基以及β-杂芳基烯酮底物同样能够良好地参与上述的反应过程,并获得相应的P–手性二级膦-硼烷产物 (2i-2n)。此外,该小组观察到,上述的标准反应体系对于各类β-烷基烯酮底物,例如异丙基(2o)、环丙基 (2p)、环己基 (2q)和杂环己基 (2r),同样能够表现出高度的对映与非对映选择性控制。接下来,作者进一步发现,α,β-不饱和N-酰基吡咯底物同样能够有效地完成上述的对映选择性转化过程,并获得相应的P–手性二级膦-硼烷分子 (2s-2u)。
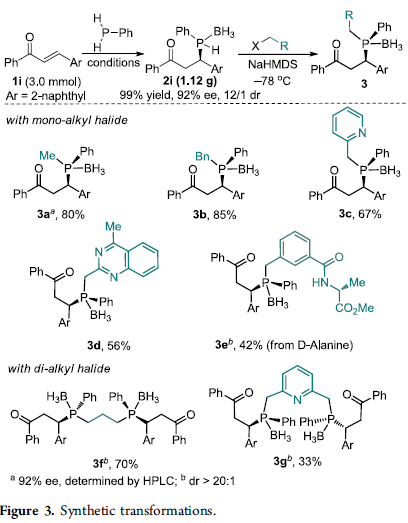
之后,该小组采用β-萘基烯酮底物,进行上述对映选择性合成转化过程的克级规模实验研究,并成功完成P-手性二级膦-硼烷产物2i的克级规模合成 (1.12 g, 99% yield, 92% ee, 12:1 dr)。同时,该小组发现,通过上述策略获得的具有P–手性二级膦-硼烷产物,能够进一步作为构建各类手性三级膦与手性双膦化合物的关键砌块。该小组在-78 oC以及 NaHMDS 存在的条件下,通过光学活性的二级膦-硼烷砌块2i对于各类卤代烷试剂的亲核进攻过程,进而以较高的反应收率,获得一系列手性三级膦 (3a–3e)与手性双膦化合物 (3f与3g),并且,反应过程中无产物光学纯度的丧失 (Figure 3)。
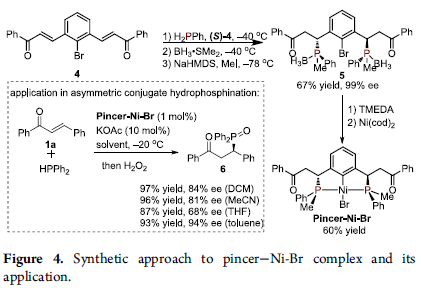
为进一步阐明上述反应策略的合成应用价值,作者进一步选择二烯酮4与苯基膦在(S)-4存在的条件下,进行相应的膦氢化过程,之后通过BH3保护以及后续的甲基亲核取代步骤,进而顺利完成具有四重立体生成中心的手性双膦分子5 (99% ee)的制备 (Figure 4)。同时,值得注意的是,手性双膦分子5为构建具有P–手性中心的Pincer-Ni-Br配合物的关键配体。并且,该小组发现,这一全新的Pincer-Ni-Br配合物同样能够进一步应用于催化不对称共轭膦氢化方法学的相关研究,并获得优良的反应收率与优良的对映选择性 (Figure 4)。
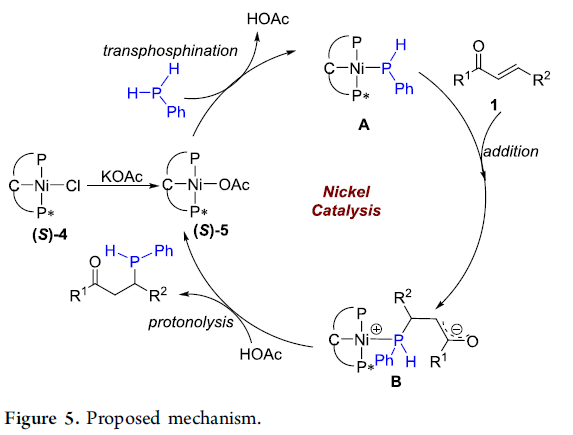
接下来,作者提出一种镍催化的H2PPh与烯酮之间不对称加成过程的催化循环 (Figure 5)。首先,通过(S)-4与乙酰氧基负离子之间的配位负离子交换过程,形成具有反应活性的PCP’-Ni-OAc配合物 (S)-5。之后,通过PCP’-Ni-OAc配合物 (S)-5与H2PPh之间的transphosphination过程,形成Ni-phosphido中间体 A (31P-NMR 谱: δ (ppm) 200.78 (d, J =247.6 Hz), 55.22 (d, J = 245.4 Hz), -57.0 (brs),详见Supporting Information)。接下来,通过A中亲核性P原子对烯酮 1中β-位置的亲核进攻过程,形成具有负离子结构单元的镍-膦配合物 B,并通过HOAc对配合物 B的质子化步骤,形成相应的膦氢化产物。同时,使活性催化剂 (S)-5再生。值得注意的是,形成的膦氢化产物在无保护基团存在时,缺乏良好的稳定性,在温度升高时,P-手性中心极易出现外消旋化,形成具有较低非对映选择性的两种异构体 (31P-NMR 谱: δ (ppm) -22 (s), -29 (s),详见Supporting Information)。
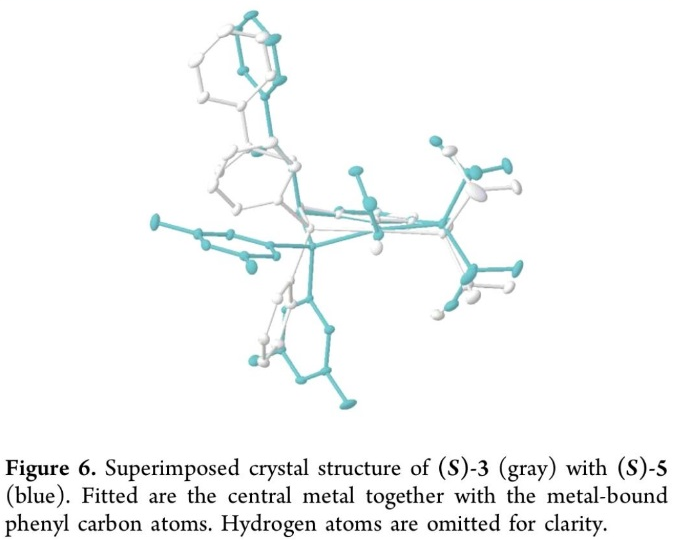
同时,作者通过XRD晶体结构分析,进一步确定出 (S)-3与 (S)-5的晶体结构 (Figure 6)。通过XRD晶体学分析,作者观察到,在(S)-3中两个磷原子几乎与通过苯基键合的镍原子位于同一平面,而在(S)-4中,两个磷原子则分布于平面两侧。这一分子结构的变形 (θ = 20.1(3)°,详见Supporting Information)源自于分子中3,5-二甲基取代的苯基基团与 -OAc之间的立体相互作用。而在Ni-phosphido中间体 A中,苯基膦配体的结构翻转则由于在(S)-4作为反应催化剂时,出现立体张力的增加而受到一定程度的限制。并且,研究发现,通过3,5-二甲基苯基代替手性镍配合物(S)-4中的苯基基团时,在反应过程中能够观察到产物对映与非对映选择性的提升,进而进一步支持立体位阻效应的存在 (Table 1, entry 4 vs entry 3)。
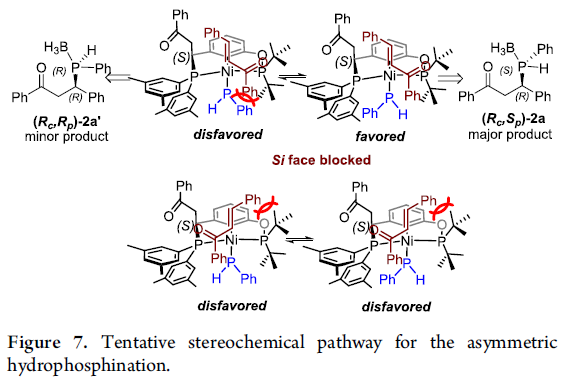
基于上述研究,作者进一步提出一种膦氢化过程中对映选择性控制的机理模型 (Figure 7)。研究表明,在形成的主要产物 2中,碳原子中心的绝对构型为(R)-构型,而磷原子中心的绝对构型为(S)-构型,这源自于Ni-phosphido中间体 A中的前手性Si面受到配位层内的非对称PCP′配体中叔丁基的有效屏蔽,进而实现膦氢化过程中,优良的对映选择性控制。
总结
综上所述,扬州大学的段伟良研究团队成功设计出一种采用非对称双膦 (PCP’) 钳型镍配合物参与的具有P-手性中心的二级膦-硼烷分子的催化对映选择性合成,并获得良好的反应收率以及优良的对映与非对映选择性。同时,这一全新的反应策略具有极为优良的官能团兼容性。此外,研究发现,相应的P-手性二级膦-硼烷砌块同样能够进一步转化为一系列有机合成方法学研究中较为关键的有机磷分子。
参考文献
[1] H. Guo, Y. Fan, Z. Sun, Y. Wu, O. Kwon, Chem. Rev. 2018, 118, 10049. doi: 10.1021/acs.chemrev.8b00081. [2] X. Du, Y. Xiao, J. Huang, Y. Zhang, Y. Duan, H. Wang, C. Shi, G. Chen, X. Zhang, Nat. Commun. 2020, 11, 3239. doi: 10.1038/s41467-020-17057-z. [3] D. Liu, B. Li, J. Chen, I. D. Gridnev, D. Yan, W. Zhang, Nat. Commun. 2020, 11, 5935. doi: 10.1038/s41467-020-19807-5. [4] G. Xu, C. H. Senanayake, W. Tang, Acc. Chem. Res. 2019, 52, 1101. doi: 10.1021/acs.accounts.9b00029. [5] D. Xu, N. Rivas-Bascón, N. M. Padial, K. W. Knouse, B. Zheng, J. C. Vantourout, M. A. Schmidt, M. D. Eastgate, P. S. Baran, J. Am. Chem. Soc. 2020, 142, 5785. doi: 10.1021/jacs.9b13898. [6] N. F. Blank, J. R. Moncarz, T. J. Brunker, C. Scriban, B. J. Anderson, O. Amir, D. S. Glueck, L. N. Zakharov, J. A. Golen, C. D. Incarvito, A. L. Rheingold, J. Am. Chem. Soc. 2007, 129, 6847. doi: 10.1021/ja070225a. [7] T. W. Chapp, D. S. Glueck, J. A. Golen, C. E. Moore, A. L. Rheingold, Organometallics 2010, 29, 378. doi: 10.1021/om900800z. [8] V. S. Chan, M. Chiu, R. G. Bergman, F. D. Toste, J. Am. Chem. Soc. 2009, 131, 6021. doi: 10.1021/ja9014887. [9] Y. Huang, Y. Li, P. Leung, T. Hayashi, J. Am. Chem. Soc. 2014, 136, 4865. doi: 10.1021/ja501007t. [10] C. Li, Q. Bian, S. Xu, W. Duan, Org. Chem. Front. 2014, 1, 541. doi: 10.1039/C4QO00017J. [11] A. Bader, T. Nullmeyers, M. Pabel, G. Salem, A. C. Willis, S. B. Wild, Inorg. Chem. 1995, 34, 384. doi: 10.1021/ic00105a058. [12] A. Ohashi, S.-I.Kikuchi, M. Yasutake, T. Imamoto, Eur. J. Org. Chem. 2002, 2535. doi: 10.1002/1099-0690(200208)2002:15<2535::AID-EJOC2535>3.0.CO;2-O. [13] A. Bader, M. Pabel, S. B. Wild, J. Chem. Soc. Chem. Commun. 1994, 30, 1405. doi: 10.1039/C39940001405. [14] O. Berger, J.-L. Montchamp, Angew. Chem. Int. Ed. 2013, 52, 11377. doi: 10.1002/anie.201306628. [15] D. Gatineau, L. Giordano, G. Buono, J. Am. Chem. Soc. 2011, 133, 10728. doi: 10.1021/ja2034816. [16] Z. Yang, D. Liu, Y. Liu, M. Sugiya, T. Imamoto, W. Zhang, Organometallics 2015, 34, 1228. doi: 10.1021/om501287k.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载
















No comments yet.