
本文作者:杉杉
导读
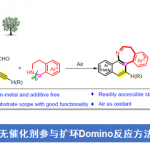
近日,有机所王鹏教授课题组在Angew. Chem. Int. Ed.上发表论文,报道了NiII催化的中性氧化还原反应(redox-neutral)以及NiII的催化循环过程。该催化循环首次实现烯烃(未活化)与苯硼酸的线性选择性氢芳化反应(hydroarylation)。值得注意的是,富电子的二亚胺配体对于反应至关重要。同时,该反应具有宽泛的底物范围、温和的反应条件、无需使用其他氧化剂和还原剂、良好的官能团耐受性等优点。此外,通过动力学分析和氘标记实验表明,质子化是该中性氧化还原反应的决速步骤,并且是新开发的二亚胺-Ni催化剂具有可逆的“链行走”(chain walking)性质。
Redox-Neutral Nickel(II) Catalysis: Hydroarylation of UnactivatedAlkenes with Arylboronic Acids
Dao-Ming Wang, Wang Feng, Yichen Wu, Tao Liu, and Peng Wang*
Angew. Chem. Int. Ed. ASAP DOI:10.1002/ange.202009195

正文
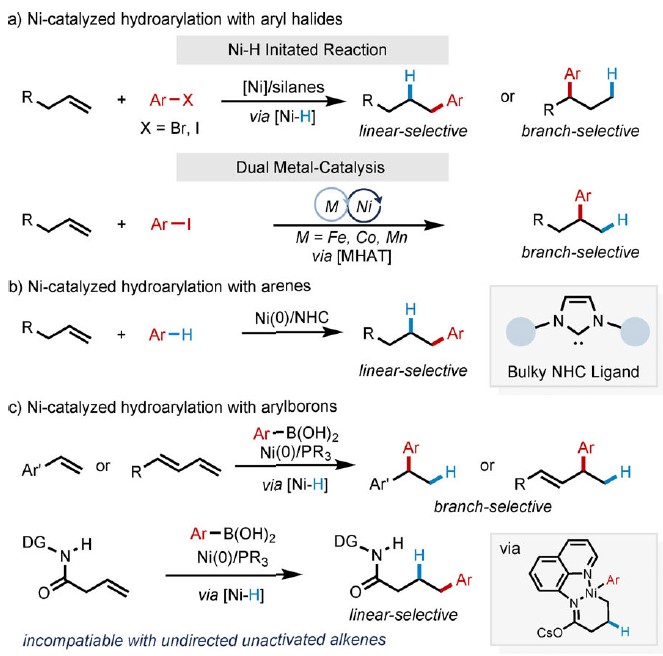
镍催化反应具有一定的优势,如多重氧化态(Ni0-NiIV)、出色的配位能力、易氧化加成和还原消除等。因此,通过双(单)电子氧化还原催化已引起了众多的关注。然而,对于具有挑战性的镍(价数不变)催化中性氧化还原反应却很少被研究。这种中性氧化还原过程可在温和条件下高效的构建碳-碳或碳-杂原子键,并具有显著的官能团耐受性,且避免氧化剂或还原剂的使用。早在1990年,Brookhart [1a]和Grubbs [1b] 提出NiII中性氧化还原工艺,涉及水杨基二氨基NiII或二亚胺NiII催化的烯烃聚合反应。在该过程中,烷基-NiII配合物能够迁移插入配位烯烃中,然后转移。然而,几乎没有将其用于烯烃官能团化中的报道。
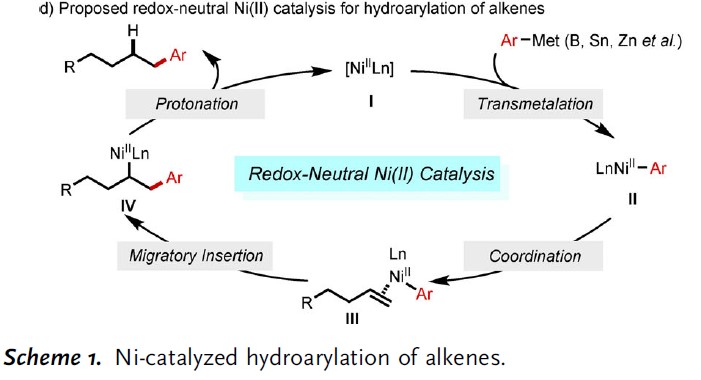
过渡金属催化烯烃与芳基的氢芳化反应是构建碳-碳键的一种有效且具吸引力的方法。在过去的几十年中,已实现多种芳基源与烯烃的区域选择性氢芳化反应。然而,由于存在一些局限性,如快速去除β-氢化物形成Hecktype副产物、原料的异构化、区域选择性难控制(特别是在无导向基团时)等,对于未活化脂肪族烯烃的直接区域选择性氢芳化反应研究相对较少。Liu [2a]和Lalic[2e]以芳基卤化物为底物,使用硅烷作为还原剂,实现了Ni催化苯乙烯和未活化烯烃的线性选择性氢芳化反应。2017年,Zhu[2c]课题组报道了通过Ni-H引发“链行走”过程,实现远程分支的选择性氢芳化反应(Scheme 1a)。Shenvi课题组[2b,2d]报道了另一种通过金属-氢原子转移对烯烃进行支链选择性氢芳化的双金属催化策略。最近,Nakao和Hartwig[3a]通过配体-配体氢转移(LLHT)机理,实现Ni0/NHC催化未活化烯烃与芳烃的线性选择性氢芳化反应,但需过量芳烃(Scheme 1b)。然而,此类体系存在一定的弊端,如还原剂或氧化剂的使用、苛刻的反应条件、复杂的催化体系、底物范围窄等。Zhou课题组[4a]研究表明,在中性氧化还原条件下,Ni0可使苯乙烯与苯硼酸发生支链选择性氢芳化反应,其中,在甲醇氧化加成后很容易产生Ni-H。后来,Zhao和Zhou [4b] 将这个概念扩展到具有线性选择性由导向基团辅助的未活化烯烃中(Scheme 1c),虽然实现了线性选择性氢芳化反应,但对于简单的脂肪族烯烃却不相容。在这篇文献中,受NiII催化的烯烃聚合过程的启发[5],作者设想,中性氧化还原过程可能适用于未活化脂肪族烯烃的氢芳化反应(Scheme 1d)。
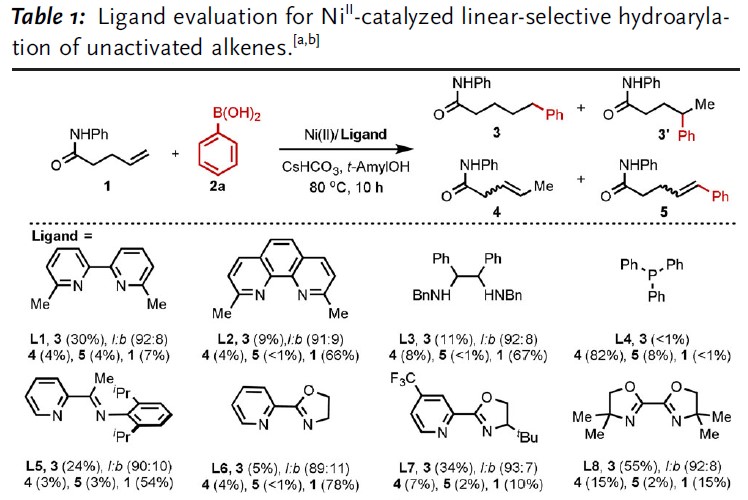
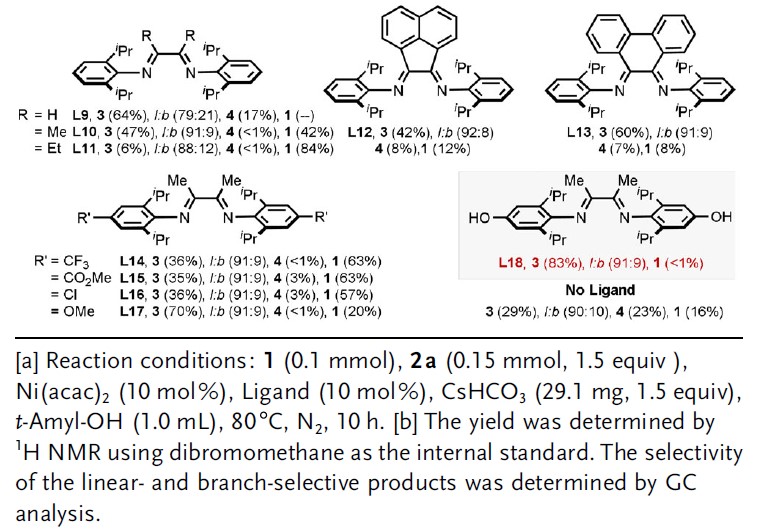
首先,作者以未活化的烯烃1和苯硼酸2a作为模型底物,进行了相关配体的筛选(Table 1)。通过线性/分支选择性表明,二亚胺配体(L9–L18)具有抑制Heck型副产物5的能力(可能是由于β-H消除反应迟缓所致)。因此,作者继续对二亚胺配体进行了修饰,最终具有富电子的对羟基取代配体L18可将收率提高至83%,线性/支链选择性为91/9。值得注意的是,假设在碱性条件下酚羟基可以转化为苯酚离子,因此提供了高度富电子的二亚胺配体,该配体能够在整个催化循环中稳定NiII催化剂。
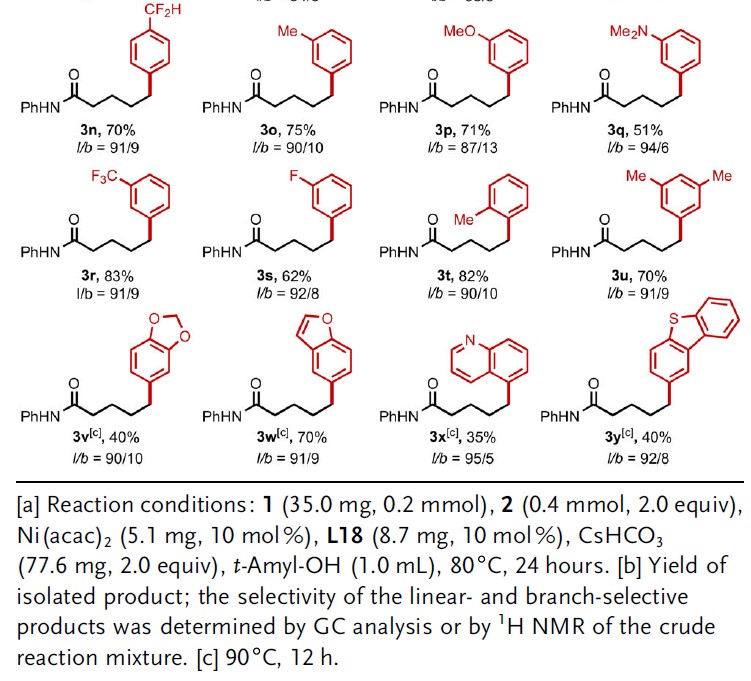
在获得上述最佳反应条件后,作者首先对苯硼酸底物进行了扩展(Table 2)。反应结果表明,苯硼酸的底物不受电子效应和定位效应的影响,均可获得相应的产物3a–3v。通常,缺电子的苯硼酸具有更高选择性。同时,具有杂环的苯硼酸底物同样与体系兼容,获得相应的产物3w–3y。

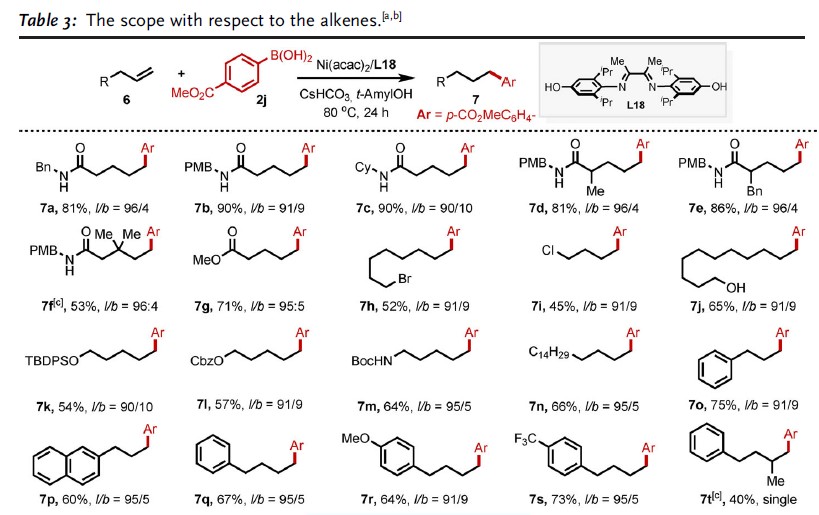
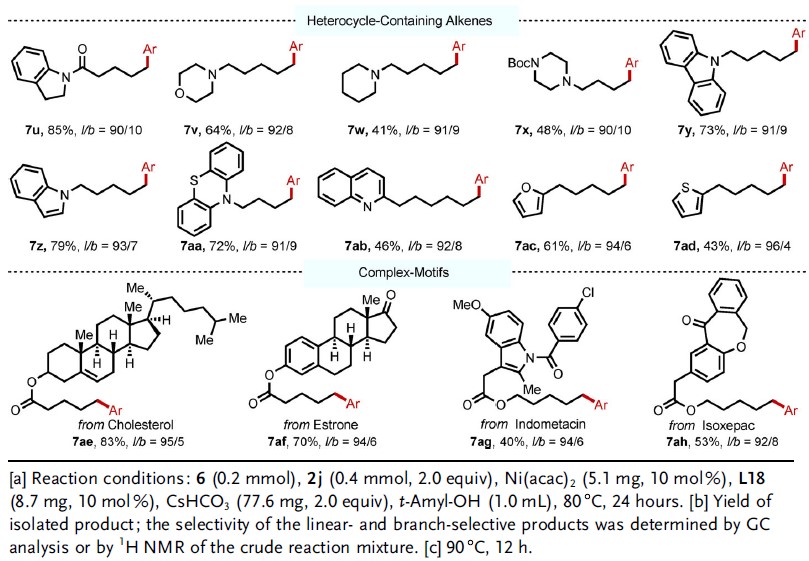
随后,作者对烯烃底物进行了扩展(Table 3)。反应结果表明,各种取代的烯烃均可与体系兼容,从而获得相应的产物7a–7t。同时,具有芳基和非芳基杂环底物,同样与体系兼容,获得相应的产物7u–7ad。此外,该方法成功的对生物分子进行了后期功能化,如胆固醇(7ae),雌酮(7af),消炎痛(7ag)和伊索克酸(7ah)。
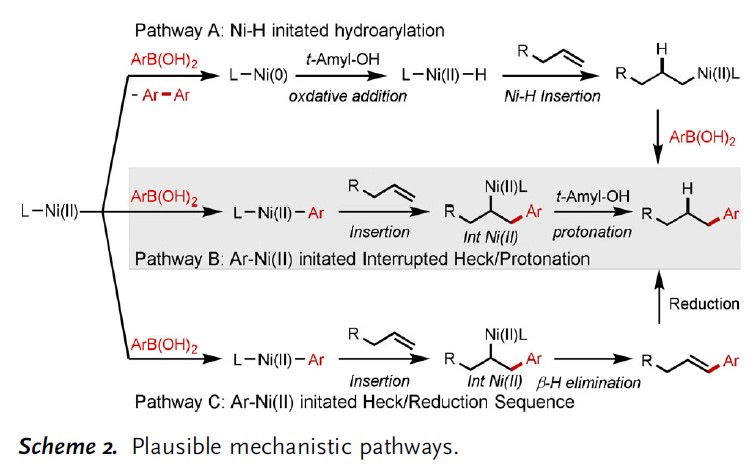
紧接着,作者提出了三种可能的途径(Scheme 2)。在Path A中,涉及苯硼酸的两次转金属化和还原消除形成Ni0,再与叔戊醇进行氧化加成以生成关键的[LNiII-H]配合物,然后迁移至未活化的烯烃中,最后经金属转移和还原消除,得到目标产物。在Path B中,通过苯硼酸的金属转移,随后中断Heck反应以形成关键的烷基-NiII中间体(Int NiII),最后该中间体经质子化获得产物并再生NiII催化剂。在Path C中,与Path B的区别在于Heck产物的形成,可以在存在Ni-H配合物的情况下进一步减少Heck产物以提供最终产物。
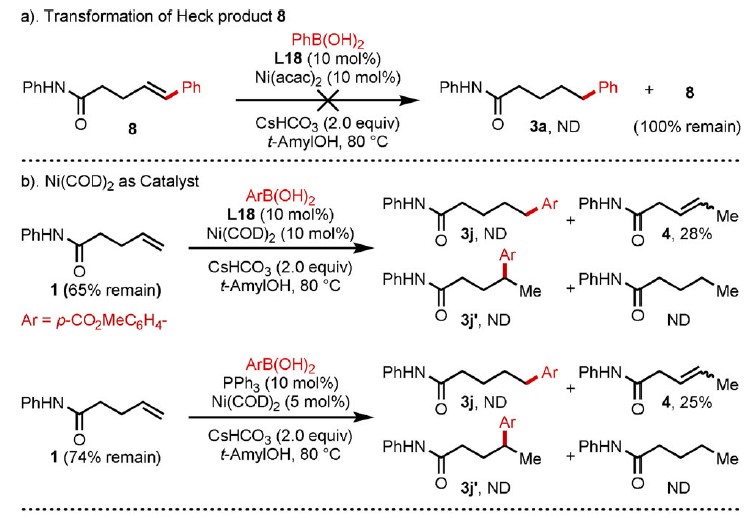
为了验证上述机理的正确性,作者进行了相关的对照实验。首先,Heck产物8在标准反应条件下未发生反应,从而排除Path C(Scheme 3a)。对于Path A-B,主要的区别在于NiII-H引发的Path A和Ar-NiII引发的Path B。使用Ni(COD)2代替Ni(acac)2导致异构化产物,而不是氢芳化产物。在标准条件下,异构化产物的形成可能表明通过醇与Ni0的氧化加成反应形成了Ni-H配合物。同样,使用Ni(COD)2/PPh3作为催化体系,得到相似的结果。然而,尽管在反应中形成了镍氢配合物,但仍未发生所需的氢芳化反应(Scheme 3b)。同时,未检测到苯硼酸的二聚和异构化副产物。这些结果表明Ni0的形成过程(Path A)不太可能适用该标准体系。
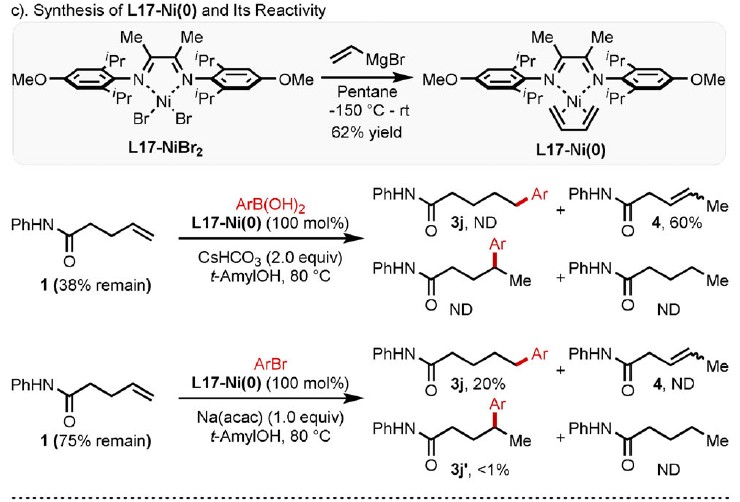
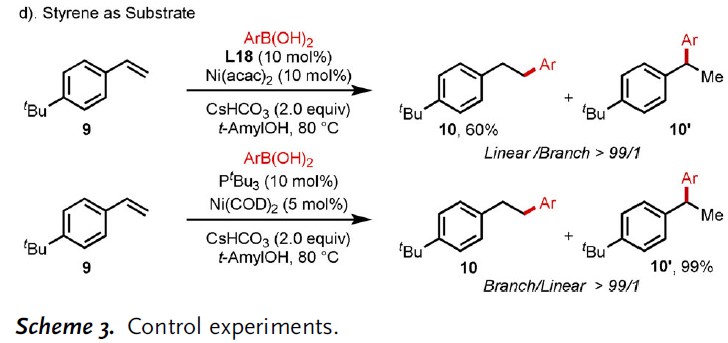
为了验证Path B的正确性,作者合成了二亚胺L17-Ni(0)配合物(Scheme 3c)。使用L17-Ni(0)配合物作为催化剂,在1H NMR中观察到60%收率的异构化产物,但仍未检测到所需的芳基化产物。然而,使用芳基溴化物作为芳基化试剂时,可获得20%收率的目标产物。从而说明,该过程通过 [L17-Ni0]配合物与芳基溴的氧化加成生成关键中间体[L17-NiII-Ar],这也可以通过在此标准反应体系中用苯硼酸进行转金属化来生成。此外,该体系还与苯乙烯型底物兼容,以60%的产率获得氢芳化产物10,并具有出色的线性选择性。若将催化体系改为Ni(COD)2/PtBu3,则以99%的收率得到了支链选择性产物。总体而言,Path B似乎更合理。同时,通过动力学分析,Int NiII中间体的质子化是限速步骤。
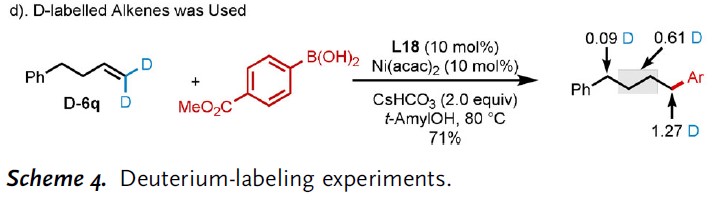
为了验证产物中的氢源,作者进行了相关的氘代实验(Scheme 4)。在标准条件下,无苯硼酸时,用氘标记的叔戊醇中处理底物1和产物3j时,未进行H/D交换(Scheme 4a)。ArB(OD)2与烯烃1反应后,得到未氘化的产物,从而排除了ArB(OD)2作为氢源的可能性(Scheme 4b)。在t-AmylOD中处理所有反应物时,发现热力学上稳定的羰基(0.15 D)和苄基位置(0.52 D)的α-位,生成部分氘化产物(Scheme 4c)。有趣的是,降低t-AmylOD中CsHCO3的负载量,会使产物中氘的贡献更高。因此,作者假设产物中的氢主要来自叔戊基羟基,部分来自碳酸氢盐。值得注意的是,氘在羰基热力学稳定位置(0.15 D)和其他位置均未观察到氘,表明二亚胺-NiII催化剂能够快速进行链行走过程(β-H消除并重新插入),并进一步证实质子化是催化循环中决速步骤。

根据上述的实验结果,作者提出了一种中性氧化还原催化循环的反应机理(Figure 1)。首先,苯硼酸与L-NiII经金属转移后,形成LNiII-Ar配合物。紧接着,插入未活化的烯烃中。最后,质子化以再生NiII并形成目标产物。此外,动力学实验表明,质子化步骤作为决速步骤。值得注意的是,新开发的镍催化剂具有快速移动特性。因此,镍的行走特性以及质子化的决速步骤,将为未活化烯烃的远程双官能化开辟新型途径。

总结
有机所王鹏教授课题组报道了NiII催化的中性氧化还原反应,首次实现烯烃(未活化)与苯硼酸的线性选择性氢芳化反应。值得注意的是,富电子的二亚胺配体对于反应至关重要。同时,该反应具有宽泛的底物范围、温和的反应条件、无需使用其他氧化剂和还原剂、良好的官能团耐受性等优点。
参考文献
- [1] a) C. Wang, S. Friedrich, T. R. Younkin, R. T. Li, R. H. Grubbs, D. A. Bansleben, M.W. Day, Organometallics 1998, 17, 3149-3151; b) L. K. Johnson, C. M. Killian, M. Brookhart, J. Am. Chem. Soc. 1995, 117, 6414-6415.
- [2] For Ni-catalyzed hydroarylation of alkenes with aryl halide, see: a) X. Lu, B. Xiao, Z. Zhang, T. Gong, W. Su, J. Yi, Y. Fu, L. Liu, Nat. Commun. 2016, 7, 11129; b) S. A. Green, J. L. M. Matos, A. Yagi, R. A. Shenvi, J. Am. Chem. Soc. 2016, 138, 12779-12782; c) F. Chen, K. Chen, Y. Zhang, Y. He, Y.-M.Wang, S. Zhu, J. Am. Chem. Soc. 2017, 139, 13929 – 13935; d) S. A. Green, S. Vasquez-C_spedes, R. A. Shenvi, J. Am. Chem. Soc. 2018, 140, 11317-11324; e) J. Nguyen, A. Chong, G. Lalic, Chem. Sci. 2019, 10, 3231-3236.
- [3] For Ni-catalyzed hydroarylation of alkenes with arenes, see: a) N. I. Saper, A. Ohgi, D. W. Small, K. Semba, Y. Nakao, J. F. Hartwig, Nat. Chem. 2020, 12, 276-283; b) Y. Schramm, M. Takeuchi, K. Semba, Y. Nakao, J. F. Hartwig, J. Am. Chem. Soc. 2015, 137, 12215-12218; c) J. S. Bair, Y. Schramm, A. G. Sergeev, E. Clot, O. Eisenstein, J. F. Hartwig, J. Am. Chem. Soc. 2014, 136, 13098-13101; d) O. Nakao, Y. Yamada, N. Kashihara, T. Hiyama, J. Am. Chem. Soc. 2010, 132, 13666-13668.
- [4] For Ni-catalyzed hydroarylation of alkenes with arylborons, see: a) L.-J. Xiao, L. Cheng, W.-M.Feng, M.-L.Li, J.-H.Xie, Q.-L. Zhou, Angew. Chem. Int. Ed. 2018, 57, 461-464; Angew.Chem. 2018, 130, 470-473; b) H. Lv, L.-J.Xiao, D. Zhao, Q.-L. Zhou, Chem. Sci. 2018, 9, 6839-6843; c) X.-Y.Lv, C. Fan, L.-J.Xiao, J.-H. Xie, Q.-L. Zhou, CCS Chem. 2019, 1, 328-334; d) Y.-G. Chen, B. Shuai, X.-T.Xu, Y.-Q.Li, Q.-L.Yang, H. Qiu, K. Zhang, P. Fang, T.-S. Mei, J. Am. Chem. Soc. 2019, 141, 3395 -3399; e) Y.He, C. Liu, L. Yu, S. Zhu, Angew. Chem. Int. Ed. 2020, 59, 9186 -9191; Angew. Chem. 2020, 132, 9271- 9276.
- [5] a) C. Wang, S. Friedrich, T. R. Younkin, R. T. Li, R. H. Grubbs, D. A. Bansleben, M.W. Day, Organometallics 1998, 17, 3149 -3151; b) L. K. Johnson, C. M. Killian, M. Brookhart, J. Am. Chem. Soc. 1995, 117, 6414 – 6415.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!












No comments yet.