
本文作者:杉杉
导读
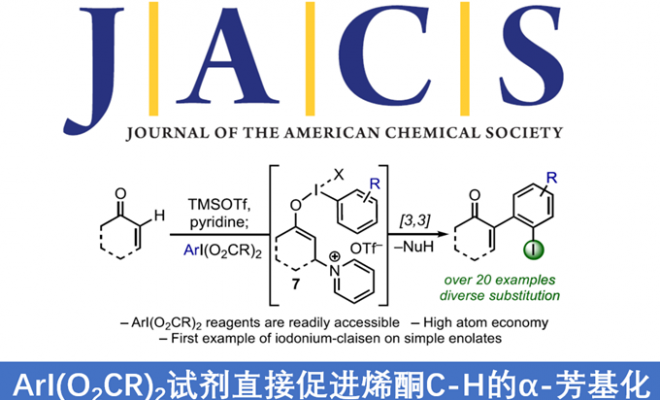
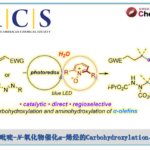
α,β-不饱和酮的α-芳基化反应作为合成中常用的手段之一,通常以α-卤代烯的交叉偶联反应来实现,但常常需要底物的预官能团化和使用昂贵的催化剂。而烯酮直接C-H的α-芳基化则体现了原子和步骤经济性的特点,但很少有文献报道。近日,天普大学Wengryniuk教授课题组在JACS发表论文,在无金属催化剂时,通过高价芳基碘试剂作为底物,实现烯酮的直接C-H芳基化反应。该反应通过原位产生的β-吡啶基甲硅烷基烯醇醚中间体7,经还原性碘鎓-Claisen重排获得α-芳基产物。产物中的芳基来自ArI(O2CCF3)2试剂,同时该反应具有较好的兼容性,可合成多种碘代芳基衍生物。机理研究表明,中间体7作为反应成功的关键,同时β-吡啶结构对于所需C-C键的形成至关重要。
Direct C–H α-Arylation of Enones with ArI(O2CR)2 Reagents
Felipe Cesar Sousa e SilvaNguyen T. VanSarah E. Wengryniuk*
J. Am. Chem. Soc. 2020, 142, 1, 64-69. DOI:10.1021/jacs.9b11282
正文
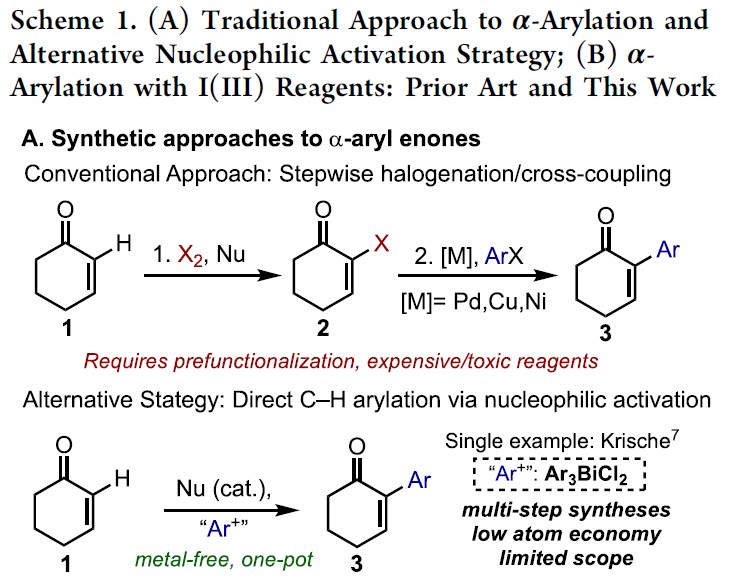
羰基化合物的α-芳基化作为一种高效构建C-C键过程,尽管过渡金属和有机催化已大量报道了关于通过烯醇盐进行酮或醛的α-芳基化反应,但相应烯酮的C(sp2)-H芳基化却很少被研究。α-芳基化反应,通常以α-卤代烯酮与芳烃底物进行偶联反应来实现(Scheme 1A),但常需对烯酮底物预官能团化处理(有时需要多步制备),并且使用昂贵或有毒的金属催化剂(如Pd,Sn)。烯酮直接C-H的α-芳基化反应,则体现了步骤与原子经济性的特点,但这种方法的开发极具挑战。2000年,Krische课题组报道了,通过亲核膦催化剂与高价铋(V)配位作为芳基转移试剂,从而实现芳基化反应(Scheme 1A)。尽管该方法未使用金属催化,同时一步实现了烯酮的C-H芳基化反应,但该方法尚未得到广泛采用,可能是由于使用不为常见的Ar3BiCl2试剂,同时需要多步合成,并且违背了原子经济性的要求(两个芳基未被合理利用)。
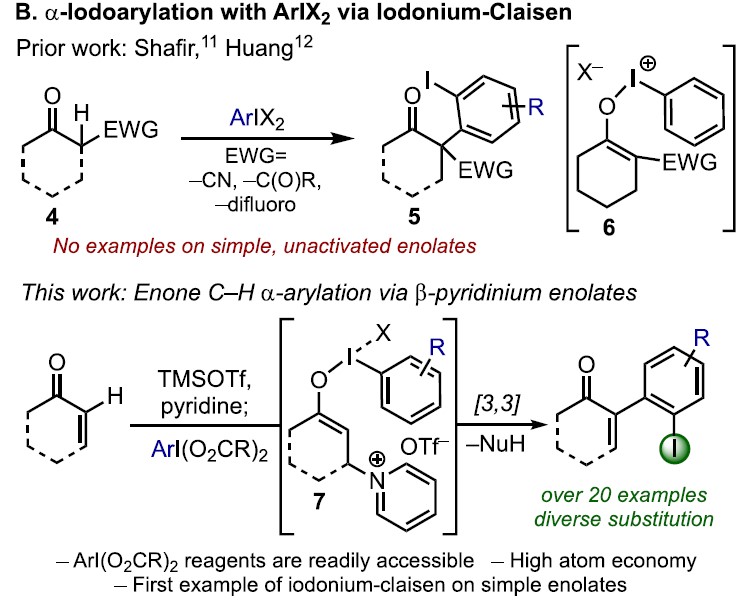
基于上述文献启发以及对高价碘化合物的持续研究,作者推测是否高价碘化合物也可作为芳基化试剂(高价碘化合物具有廉价易得、易于处理、应用广泛等优点)。在I(III)物种中,二芳基碘鎓盐[Ar2IX]已广泛应用于芳基转移试剂,但存在原子经济性低以及非对称化学选择性的问题。Shafir等报道中,证明了ArIX2试剂可以影响烯醇化合物(4)的芳基化,包括1,3-二羰基,α-氰基酮和 α,α-二氟甲硅烷基烯醇醚,通过与O-I结合的烯酸酯的还原性碘鎓-克莱森重排(6)(Scheme 1B)。使用ArIX2作为芳基化试剂具有明显的优势,因为它们很易从芳烃碘母体中获得,同时具有较高的原子经济性。然而,迄今为止,尚无成功的芳基化合物实现[3,3]烯醇盐或烯醇醚上的碘鎓-克莱森重排。在此,作者通过原位生成的β-吡啶基甲硅烷基烯醇醚(7)和ArI(O2CCF3)2试剂,经还原碘鎓-克莱森重排,实现烯酮直接C-H的α-芳基化(Scheme 1B)。
而这类反应中,最大的挑战是控制对所需C-X键的形成(Scheme 2A)。两种产物都可能来自相同的中间体9,该中间体在α-碳上具有亲电性,易受亲核试剂的进攻。通过还原性的[3,3]重排实现α-芳基化反应(path a),但取代的X配体发生选择性分子间进攻,则获得α-氧化产物(path b)。基于这两种可能的机制,作者假设[ArI(het)2] X2或N-HVIs可能非常适合此转化,因为杂环氮配体具有较弱结合且相对不亲核性,因此有利于目标产物10的形成。
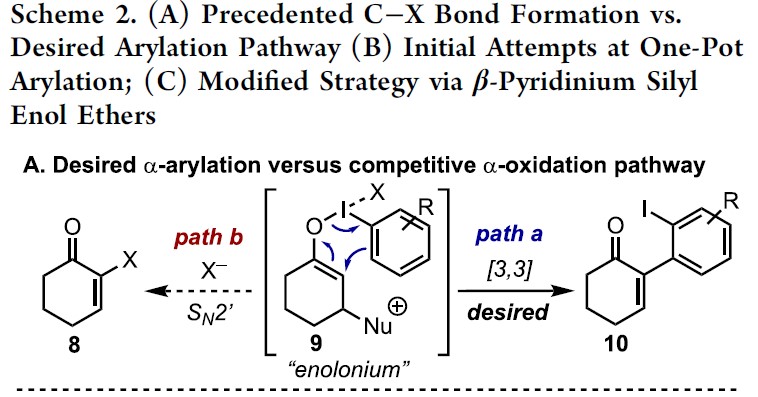
使用环己烯酮作为模型底物时,使用吡啶作为外源性亲核试剂时,促使吡啶相连的N-HVI [PhI(Py)2](OTf)2反应获得收率23%的α-碘代芳基烯酮12(Scheme 2B )。遗憾的是,在筛选了N-HVI、其他I(III)试剂、β-亲核试剂和反应条件时,均无法使芳基化反应产率提高。作者推测,可能是由于I(III)试剂与亲核试剂之间竞争性导致较差的结果(如配体交换、氧化降解、配体偶联等)。为了解决这个问题,作者提出了一步法的芳基化过程,其中将ArIX2加入到原位形成的烯酸酯中。此外,根据Kim报道文献的总结,作者使用TMSOTf和吡啶作为反应条件,通过1H NMR分析从而证明了β-吡啶基甲硅烷基烯醇醚13的完全转化(Scheme 2C)。
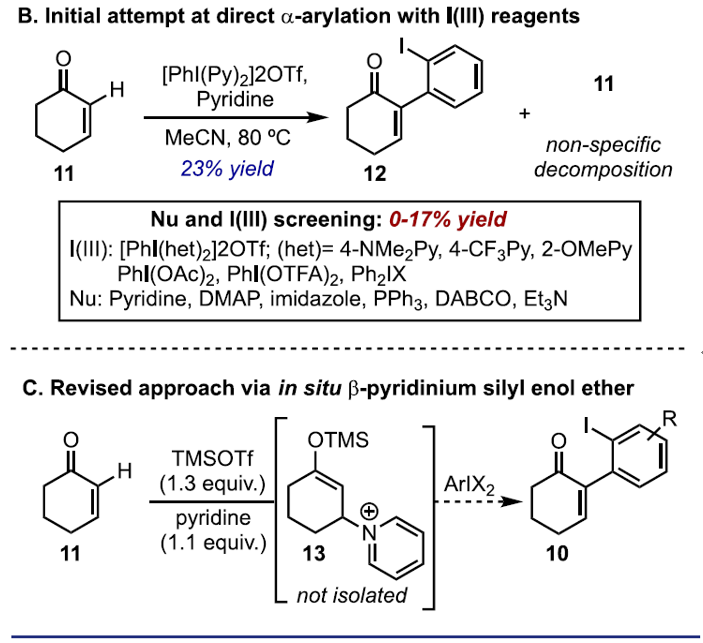
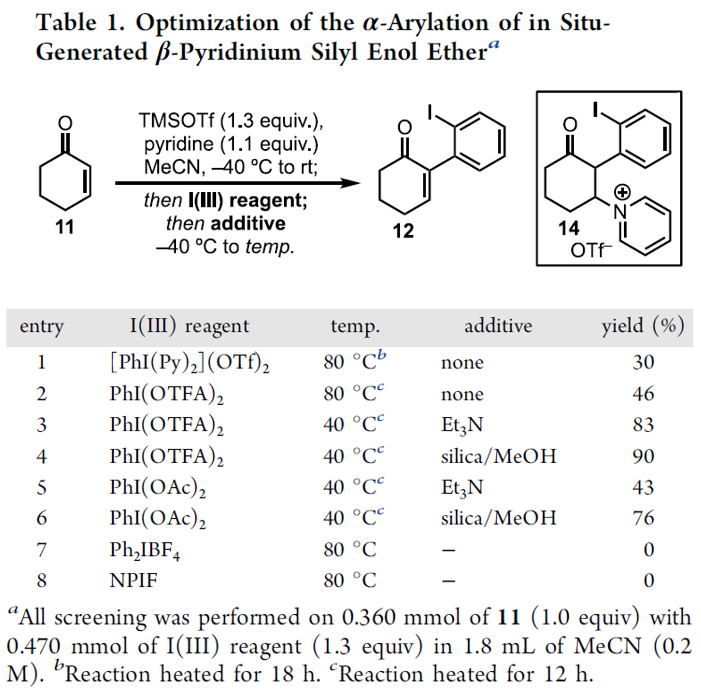
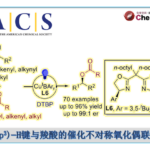
首先,作者以11作为模型底物,在上述条件下获得β-吡啶基甲硅烷基烯醇醚13后,对I(III)试剂与添加剂进行了筛选(Table 1)。当以[PhI(Py)2](OTf)2作为芳基碘试剂时,加热至80℃反应18 h,获得30%收率的α-芳基化产物12(entry 1)。而使用PhI(OTFA)2试剂时,反应12h可将收率提高至46%(entry 2)。有趣的是,并没有发现α-氧化副产物,通过NMR分析可知,反应中存在芳基化吡啶鎓盐中间体14(分离不稳定),同时在低温下α-芳基化迅速发生(<10min),而14至12的转化则为缓慢过程。根据这一发现,作者对各种添加剂进行了研究,以加快β-吡啶盐的消除。在40℃时,添加NEt3可使收率显着提高至83%(entry 3),而使用酸性二氧化硅/ MeOH可进一步将收率提至90%(entry 4)。而将芳基碘试剂改为PhI(OAc)2时,无论加入碱性和酸性添加剂均使收率有所下降(entries 5-6)。如果使用其它二芳基碘鎓盐进行芳基化时(entries 7-8),均未反应,进一步说明了β-吡啶鎓类化合物对该反应的重要性。
在获得上述最佳反应条件后,作者开始对底物烯酮与进行了扩展(Table 2)。首先,作者对芳基碘试剂进行了扩展,芳基碘化物不受定位效应影响,均可获得相应的α-(2-碘代芳基)烯酮15–19。而对电子效应的研究表明(15–24),吸电子基常常导致较低的收率,如-NO2、-CN等。同时,一些在邻位、间位和对位含有卤素的底物也被研究,因为这些产物可进行多种后期修饰,并且也为传统的金属催化偶联反应提供了选择性偶联底物。虽然均可获得相应的芳基化产物(25–33),但收率和取代位置之间出现了明显的趋势(对>间≫邻)。同时,多取代的芳烃(34、35),富电子的杂芳族化合物(36、37),也同样实现了芳基化反应。而对烯酮底物扩展时,环戊烯酮(38)和无环巴豆醛(40)的芳基化收率较高,而使用环庚烯酮(39)仅获得12%的收率。

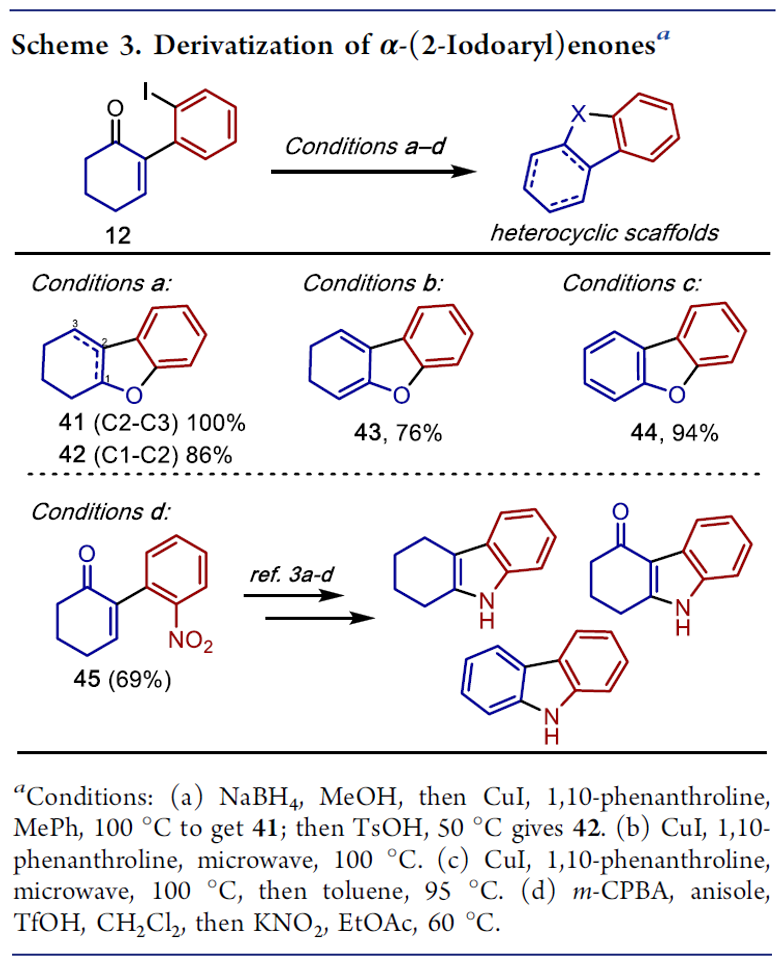
随后,作者开始对产物α-(2-碘芳基)烯酮进行了后期修饰,可有效的构建多环杂芳烃(Scheme 3)。利用羰基氧与碘的偶联可获得优异收率的苯并呋喃衍生物(41–44)。 同时吲哚类骨架已广泛应用于天然产物的合成中,而以硝基衍生物45作为底物时,可通过相关方法合成多种吲哚类衍生物。
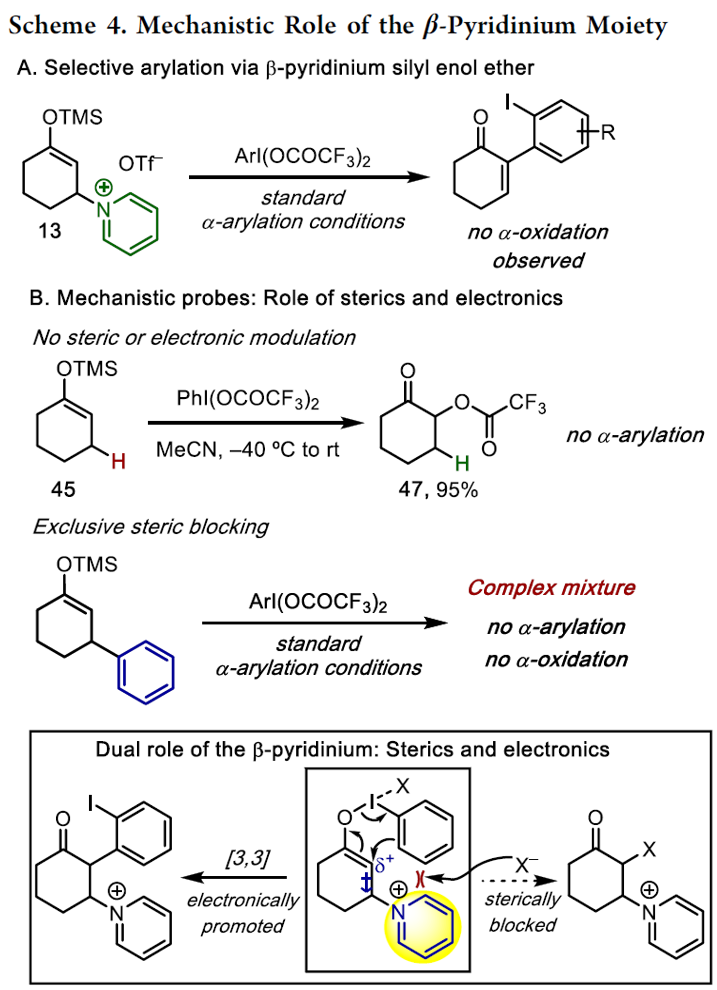
为了进一步了解反应机理,作者进行了相关的对照实验(Scheme 4)。在使用β-吡啶盐中间体13时,在形成的芳基化过程中并没有观察到α-氧化副产物(Scheme 4A)。此外,将β-未取代的甲硅烷基烯醇醚45用PhI(OCOCF3)2处理,获得等量的α-氧化产物47,说明在没有任何烯醇化物时仅获得α-氧化物(Scheme 4B)。接下来,为了消除电子效应的影响,以β-苯基甲硅烷基烯醇醚为底物在上述标准条件反应(Scheme 4B)。有趣的是,获得产物降解的混合物,同时无α-氧化或α-芳基化产物。该结果表明,单独的苯基不足以实现有效的α-芳基化,但确实起到抑制分子间的作用。综上所述,β-吡啶基中间体可能通过空间与电子效应的相互作用来调节反应过程(Scheme 4 inset)。
总结
本文主要介绍了一种通过ArI(O2CCF3)2试剂直接进行烯酮C-H的α-芳基化反应。该反应通过原位产生β-吡啶基甲硅烷基烯醇醚中间体7,然后进行还原性[3,3]重排,获得多种α-芳基化衍生物,同时作为第一个碘鎓-克莱森重排的例子。通过将α-(2-碘芳基)烯酮后期修饰,合成多种具有价值的杂环骨架,进一步证明了该方法的实用性。机理研究表明,β-吡啶基中间体的空间与电子效应对于反应至关重要。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!















No comments yet.