
本文作者:杉杉
导读
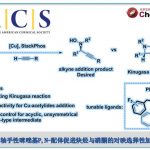
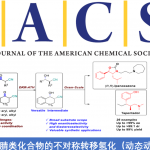
近日,香港中文大学Tsui Gavin Chit教授课题组在德国应化杂志(Angewandte Chemie-International Edition)发表论文,报道了在Pd(PPh3)4催化下,实现四取代二氟烯烃与末端炔烃的立体选择性C-F键炔基化反应,获得多种共轭单氟炔基衍生物,同时还带有立体选择性的四取代烯烃部分。此外,螯合的Pd氧化加成到C-F键中,作为立体选择性控制的关键之处。同时,通过关键的单氟乙烯基Pd(II)中间体的X-射线结构分析,进一步证明了机理的正确性。
Stereoselective Palladium-Catalyzed C-F Bond Alkynylation of Tetrasubstituted gem-Difluoroalkenes
Qiao Ma, Yanhui Wang and Gavin Chit Tsui
Angew. Chem. Int. Ed. ASAP DOI: 10.1002/anie.202002219

正文
碳-氟键的活化,一直作为有机氟化学研究的热门领域,而通过选择性对多氟或全氟化合物中C-F键断裂和功能化,则具有重要的意义。如,通过二氟烯烃化合物将其中一个C-F键选择性的官能团化后,则可获得单氟烯烃衍生物。同时,一氟烯烃衍生物广泛存在各类药物中,以及作为有机合成中氟化反应的合成子(Scheme 1a)。据文献报道,一些过渡金属催化(Cu、Ni、Rh、 Co、Mn、Ru、 Ir、Fe)已实现氟代烯烃C-F键的官能团化反应。这些反应中,通过高立体选择性的控制,实现单个C-F键功能化。通常,有两个因素可以促进选择性的控制:(1)使用可由相应的醛制备三取代的二氟烯烃化合物;(2)在β-F消除步骤中,R1和FG之间存在空间排斥(Scheme 1b)。但是,对于不对称的四取代二氟烯烃化合物(Scheme 1c)会阻碍立体选择性,特别是当R1和R2具有相似的空间特性时,会导致形成A和B的异构体,从而很难通过常规方法进行分离。因此,作者假设是否可以通过金属催化,实现C-F键的选择性氧化加成,从而获得四取代单氟烯烃衍生物。迄今为止,尚未有文献报道此类方法,可能是由于强的C-F键能(脂族和烯烃C-F键为120-129 kcal/mol)以及难以确定合适的底物和催化体系。在此,香港中文大学Tsui Gavin Chit教授课题组报道了第一个Pd催化体系,实现二氟烯烃化合物与炔烃的选择性C-F键炔基化反应,涉及C-F键氧化加成的关键步骤,同时可以合成四取代的单氟炔烃衍生物(共轭的1,3-烯炔类衍生物)。

钯催化剂作为交叉偶联反应中最强大的催化剂之一,但对于钯催化二氟烯烃的脱氟偶联反应却很少被研究。Heitz课题组早在1991年首次报道了Pd催化的1,1-二氟乙烯与碘代芳烃的偶联反应。直到2016年,Toste课题组报道了Pd催化的二氟苯乙烯与芳基硼酸的立体选择性偶联反应。此类反应,均将二氟烯烃插入钯-芳基键中,然后经β-F消除获得单氟烯烃产物。虽然对于Pd催化二氟烯烃C-F键的氧化加成过程未被提及,但Ogoshi课题组在Pd催化的四氟乙烯(TFE)与芳基锌试剂、芳基硼酸酯和芳基硅氧烷的偶联反应中,提供了关键的三氟乙烯基钯配合物的分离和X-射线晶体的重要数据,从而提出了涉及Pd氧化加成到C-F键中,然后与有机金属试剂进行金属转移的反应机理。尽管这些Pd催化的反应可以形成氟代烯烃的Csp2-Csp2键,但相应的Csp2-Csp键仍然未知。此外,作者发现,在催化Pd(PPh3)4和NaI的存在时,可实现二氟丙烯酸酯衍生物1a与末端炔烃2a的Sonogashira脱氟偶联反应,从而获得四取代单氟炔烃产物(E)-3a,产率为87%,并且通过19F-1H HOESY NMR实验证实了烯烃的几何构型的正确性。
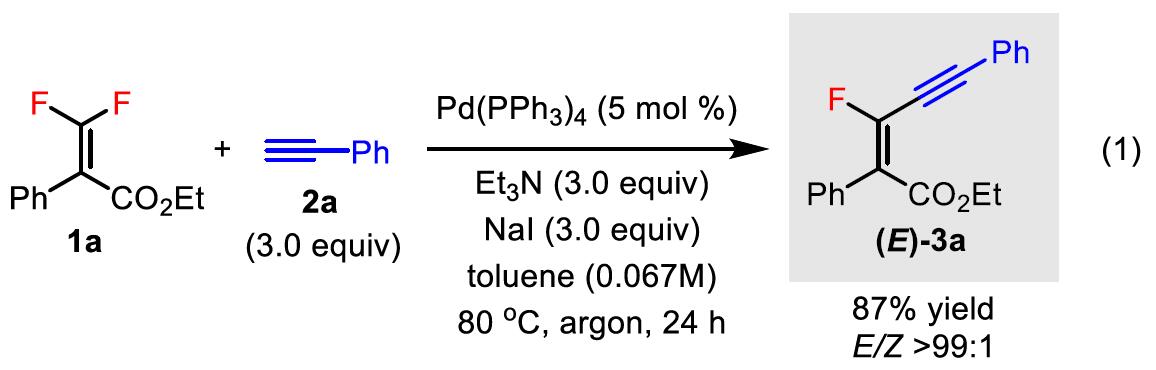
首先,作者通过大量条件的筛选之后,进行了相关的总结:(1)钯催化剂与膦配体对于反应至关重要。如PdCl2(PPh3)2、PdCl2(dppf)·CH2Cl2的Pd(II)催化剂也有效,但对于其它的Pd(II)催化剂,如PdCl2、PdCl2(CH3CN)2和Pd(OAc)2,需要添加PPh3(Pd:P=1:2)才能顺利反应。Pd(PPh3)4因其操作简单(未添加配体)和高反应活性最终作为最佳催化剂。(2)碘化钠对反应性和选择性均至关重要。若在标准条件下不添加NaI时,会导致收率低(41%)。与NaI相比,使用NaBr、NaCl和KI作为添加剂时,收率均有所下降。同时,作者对可溶性更强的四丁基卤化铵进行了研究,发现阴离子效应(I–>Br–>Cl–)对于E/Z选择性具有重要的影响(如TBAI、TBAB和TBAC,E/Z分别> 99:1、94:6和91:9)。(3)为了完全转化,需要更多的稀释浓度(0.067 M)和过量的碱和炔烃(各自为3.0 eq)。(4)使用具有不同极性的溶剂(如1,4-二恶烷、DMF)也可获得良好的产率和选择性。(5)在较低的温度下(23-60℃),即使增加了催化剂的负载量并延长了反应时间,反应仍然缓慢。(6)在某些情况下,Pd(PPh3)4的负载量可低至2 mol%,产率不受影响。
在获得上述最佳反应条件后,作者开始对底物二氟烯烃1与炔烃2进行了扩展(Scheme 2)。首先,作者固定二氟烯烃底物1,对末端炔烃2的R3取代基进行了研究(可获得产物3b–3u)。通常,富电子芳烃的收率要高于缺电子芳烃(如3c与3g–3i比较)。但使用溴取代的化合物时,产物3m的收率偏低。含有噻吩(3o)、吡啶(3p)取代基的杂环化合物,同样与体系兼容。同时,TMS-取代的化合物(3q)仅使用2mol%的Pd催化剂,即可完成克级实验。此外,带有伯碳、仲碳和叔碳的烷基取代的产物(3r–3t)也可与体系兼容,并且该方案可对天然产物的衍生物3u进行相关的修饰。
随后,作者对二氟烯烃底物1中的乙烯基取代基(R1)和酯取代基(R2)的反应范围进行了研究(产物3v–31a)。具有吸电子的芳基取代基(4-CF3,3x)比给电子的基团(4-OMe,3w)具有更高的收率。芳基氯化物(3ac,3ad)和溴化物(3ae),同样可进行Sonogashira偶联反应,从而证明C(乙烯基)-F键功能化的优先级高于C(芳基)-Cl/Br键。同时,含有萘基(3af)和噻吩基(3ag)的取代基也可获得满意的结果。紧接着,作者对R2取代基的底物范围也进行了扩展(获得产物3y–3ab)。反应结果表明,空间位阻影响反应的收率,如乙基(3y)、苄基(3z)和异丙基(3aa)的收率相近,但叔丁基(3ab)的收率大幅下降。此外,通过联苯产物3ah的X-射线晶体结构,进一步确认了产物构型的正确性。含有腈(3ai)和甲酯(3aj)的联苯底物也可与体系兼容。而在标准条件下,苄基取代的二氟烯烃反应性不如(杂)芳基对应的底物。但是,适当增加Pd的催化量(10 mol%)和改变碱(DIPEA)可以显着提高产率(3ak),甚至含有原酸酯基团也可以耐受(3al)。最后,使用1,4-二乙炔基苯与2.0当量的1a反应,可获得一个共轭的产物3am(带有两个单氟炔基单元的单一异构体)。
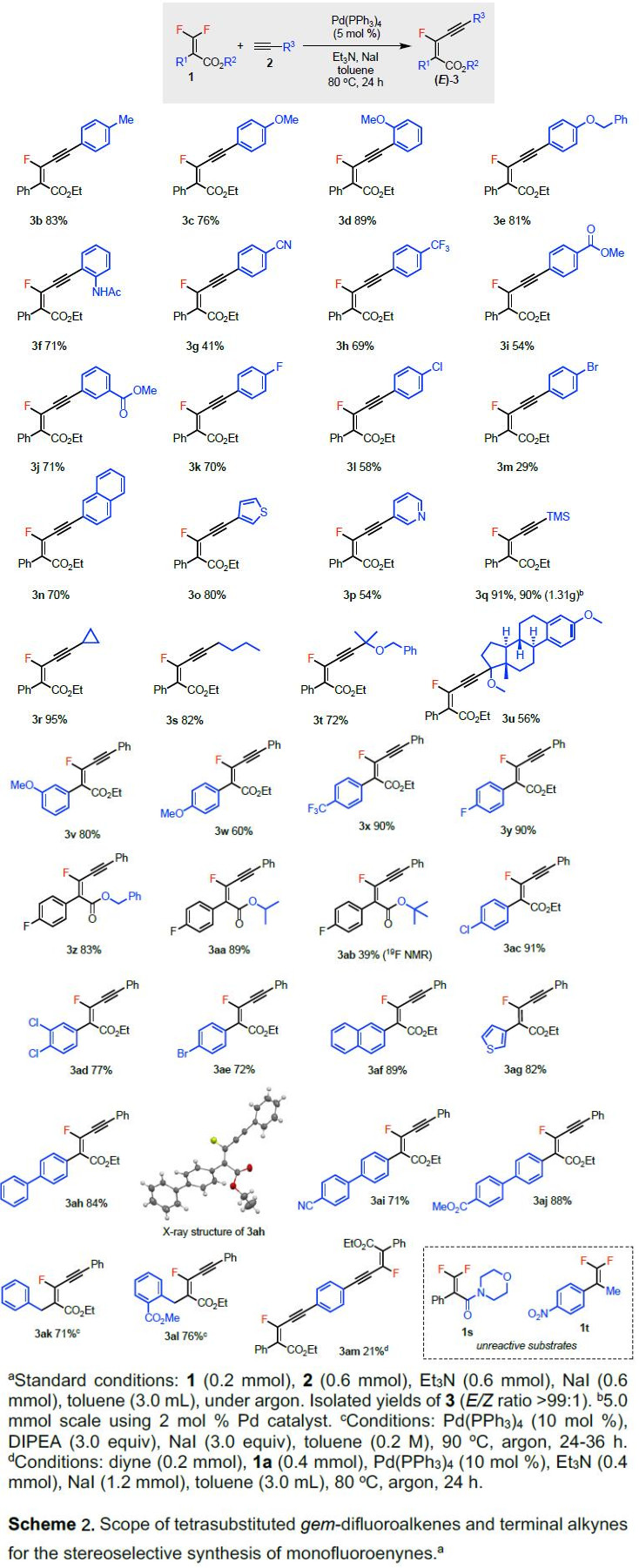
为了进一步了解反应的机理,作者进行了相关的对照实验(Scheme 3)。当1a与Pd(PPh3)4在NaI存在下进行反应时,作者进行相关的分离,从而获得一种新型的单氟乙烯基碘化钯(II)中间体Int-1,并通过X-射线晶体结构,进一步表明了Pd与两个PPh3配体之间存在反式结构(Scheme 3a),作为衍生自双二氟烯烃的钯络合物的首次报道。此外,Pd配合物Int-1与末端炔烃2a反应,可以获得高收率的产物(E)–3a,证明了Int-1作为该催化循环中关键性的中间体(Scheme 3b)。同时,在标准条件下,使用催化量的Int-1,也可获得较高收率的 (E)–3a,证明了Int-1具有一定的催化循环能力(Scheme 3c)。
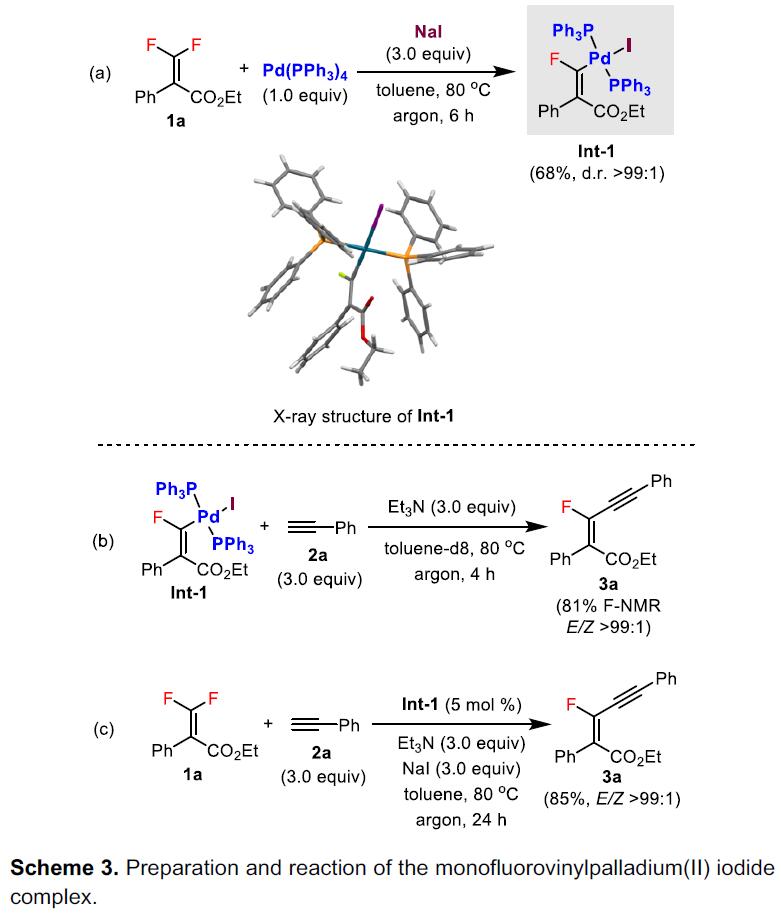
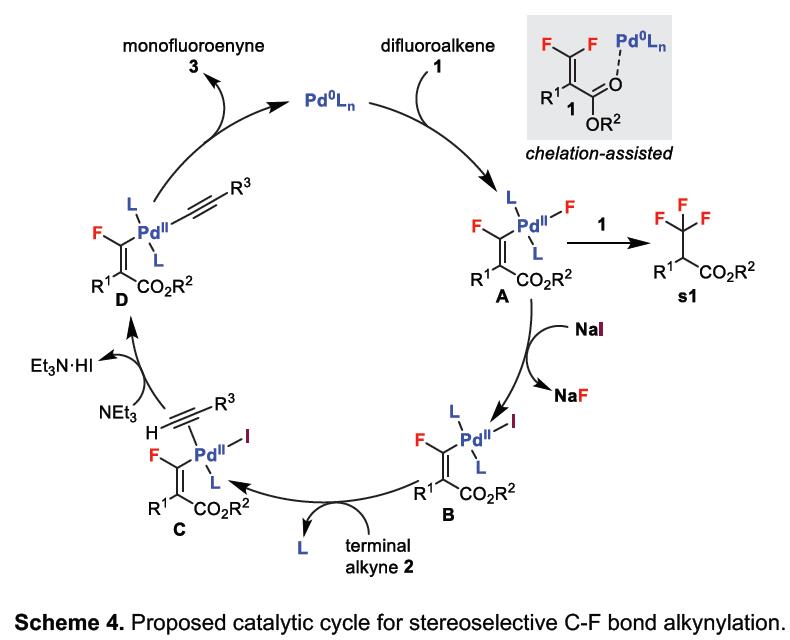
根据上述的实验和相关文献的查阅,作者提出了一种可能的反应机理(Scheme 4)。首先,将Pd(0)氧化加成至二氟烯烃化合物1的C-F键中,从而选择性地生成R-Pd(II)-F配合物A。尽管配合物A能够与末端炔烃进行反应,并在一定程度上生成所需的产物,但它也可进行迁移插入,释放氟原子,形成主要的副产物s1。但是,配合物A可与NaI中的卤化物进行交换从而抑制副反应的发生,生成R-Pd(II)-I配合物B,并消除了NaF。NaF的消除实际上可能是C-F键断裂的驱动力,因为NaF键较强,而NaF在溶剂体系中的溶解性较差。随后,末端炔烃2与Pd中心配位形成配合物C。此外,由于配位而使炔2中的质子酸度增加,可通过弱碱(例如NEt3)进行去质子化,然后与碘化物进行配体交换(整体消除了HI分子),从而得到R-Pd(II)-炔烃D。最后,经还原消除获得单氟炔基产物3,并再生Pd(0)以完成催化循环。
总结
香港中文大学Tsui Gavin Chit教授课题组报道了,在Pd(PPh3)4催化下,实现四取代二氟烯烃与末端炔烃的立体选择性C-F键炔基化反应,获得多种共轭单氟炔基衍生物。该反应具有广泛的底物范围,不受电子效应和定位效应的影响,可耐受多种官能团,如杂芳基、萘基等,同时,该反应条件下体现了C(乙烯基)-F键功能化的优先级高于C(芳基)-Cl/Br键。此外,螯合的Pd氧化加成到C-F键中,作为立体选择性控制的关键之处,并且通过关键的单氟乙烯基碘化钯(II)中间体的X-射线结构分析,进一步证明了机理的正确性。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!













No comments yet.