
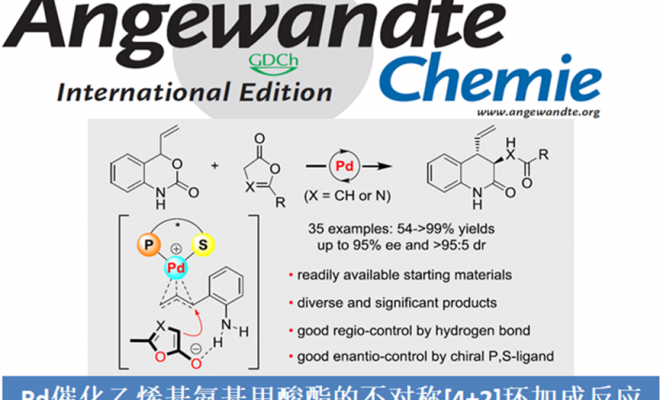
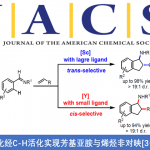
丁烯酸内酯及其衍生物广泛存在于具有生物活性的化合物中,因此丁烯酸内酯成为化学团体关注的重中之重。在过去的几十年里,关于丁烯酸内酯的不对称催化已经取得了不错的进展,其中表现较为突出的是有机催化的不对称加成反应。但有机催化丁烯酸内酯的不对称反应主要局限于γ-加成,关于丁烯酸内酯α-加成的报道还比较少[1]。过渡金属催化与有机催化同样作为合成复杂分子强有力的合成方法,但与有机催化相比,过渡金属催化丁烯酸内酯的反应只有2009年报道的Pd催化丁烯酸内酯的γ-芳基化反应这一例[2]。因此,开发过渡金属催化丁烯酸内酯的反应尤为迫切(Scheme 1a)。华中师范大学陆良秋和肖文精团队开发了手性杂化P,S络合物催化乙烯基氨基甲酸酯和亲电偶极子的不对称[4+2]环加成反应(Scheme 1b)。紧接着,作者又利用这些配体实现了Pd催化的乙烯基氨基甲酸酯和亲核偶极子的不对称[4+2]环加成反应(Scheme 1c)。基于之前的工作,华中师范大学陆良秋和肖文精团队报道了第一例Pd和新型P,S-杂化配体催化乙烯基氨基甲酸酯与丁烯酸内酯或二氢唑酮的不对称[4+2]环加成反应。其中,重庆大学的蓝宇教授课题组提供了计算化学支持。作者推测可能的机理:手性P,S-杂化配体和Pd(0)催化剂与乙烯基氨基甲酸酯1发生氧化加成反应生成含Pd的1,4-偶极子中间体A。然后,A与烯醇化的丁烯酸内酯2发生不对称烯丙基烷基化反应生成中间体B。紧接着,B会转化为中间体C,同时,Pd(0)催化剂再生。C发生分子间重排反应生成产物3。相关研究成果发表于
Inverse Electron-Demand, Pd-Catalyzed Asymmetric [4+2] Cycloadditions Enabled by Chiral P,S-Ligand and Hydrogen Bonding
Wang, Y.-N.; Xiong, Q.; Lu, L.-Q.;* Zhang, Q.-L.; Wang, Y.; Lan, Y.;* Xiao, W.-J.* Angew. Chem. Int. Ed. 2019, early view
DOI: 10.1002/anie.201905993
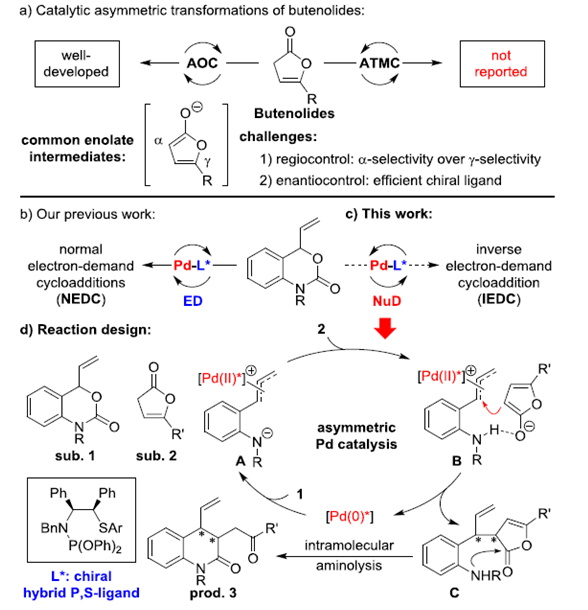
Scheme 1.研究背景
论文作者介绍:

研究者:肖文精教授,化学学院院长
教育与研究经历
- 1980.09-1984.06,华中师范大学,化学系,学士学位
- 1984.07-1990.03,华中师范大学农药化学研究所,助教
- 1987.09-1990.06,华中师范大学,农药化学研究所,有机化学专业,理学硕士学位;
- 1990.04-1994.09,华中师范大学农药化学研究所,讲师
- 1994.10-1996.03,华中师范大学农药化学研究所,副教授
- 1995.09-1996.01,中国科学院上海有机化学研究所有机化学专业博士研究生;
- 1996.03-1997.08,加拿大Ottawa大学化学系访问教授;
- 1997.09-2000.12,加拿大Ottawa大学化学系研究生学习,获理学博士学位;
- 2001.01-2002.01,美国加州理工学院化学化工部博士后研究;
- 2002.02- 2003.08,美国Materia, Inc.研究科学家、Group Leader;
- 2003.09至目前,华中师范大学化学学院,教授、博士生导师;
获奖经历
- 2010,华中师范大学第五届感动校园人物
- 2010,华中师范大学2008-2010年度“三育人”先进个人、标兵
- 2010,湖北省新侨创业先进个人
- 2010,湖北省归侨侨眷先进个人
- 2010,湖北省第五届优秀科技工作者
- 2011,Best Poster Presentation Award, 16th IUPAC International Symposiumon Organometallic Chemistry
- 2011,湖北省高等学校“优秀共产党员”称号
- 2011,全国教育系统职业道德建设标兵
- 2013,全国百篇优秀博士学位论文指导教师
- 2013,湖北省自然科学一等奖,获奖项目:碳/杂环高效构筑中的新试剂、新催化体系和新反应研究
- 2013,第七届“药明康德生命化学研究奖”,获奖项目:杂环合成中的串联反应策略
- 2014,Asian Core Program Lectureship Award, Singapore
- 2014,国家有突出贡献中青年专家,人社部
- 2014,国家百千万人才工程国家级人选,人社部
- 2015,湖北省先进工作者,湖北省人民政府
研究领域与兴趣
- 可见光促进的光氧化还原催化的可控自由基反应及其合成应用;
- 基于仿生合成策略、利用弱酸-弱碱的兼容性,发展硫叶立德与缺电子组分的新反应以及高效构筑稠杂环的新反应试剂;
- 发展新型的、结构和活性易调的双功能二级胺–氢键催化体系,实现碳/杂环体系的对映选择性去对称化;
- 基于吲哚的串联反应设计,实现稠、螺环吲哚衍生物的不对称合成
论文概要:
以乙烯基氨基甲酸酯1a和丁烯酸内酯2a为模板底物,作者对反应条件进行反复筛选,确定最优条件(Table 1):5 mol% Pd2(dba)3·CHCl3和12 mol% 手性配体L6为催化剂,DCE为溶剂,在0℃条件下反应20 h,然后升至室温,能以92%的收率和93%的对映选择性得到相应产物。
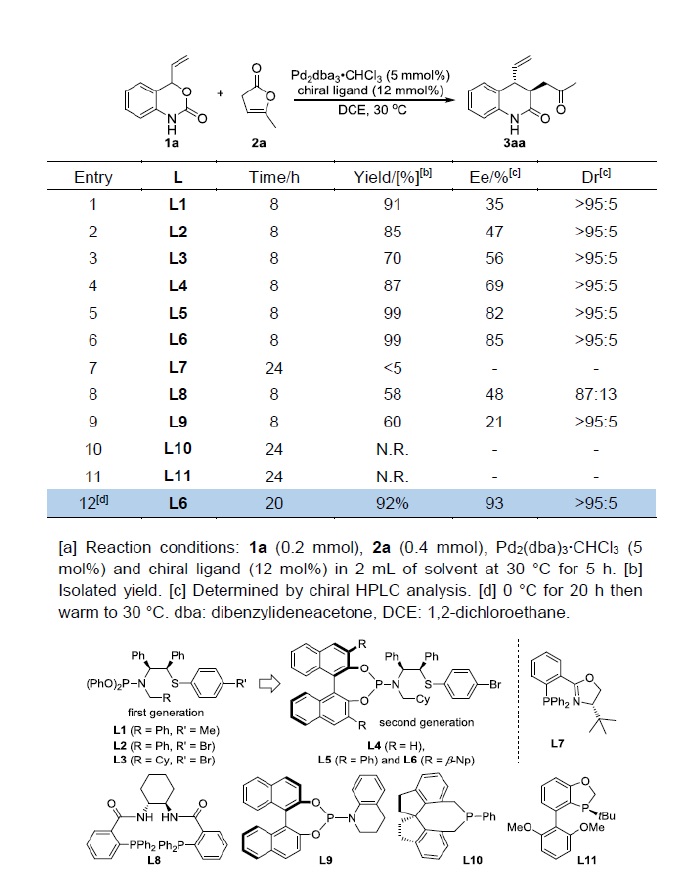
Table 1.条件优化
在最优反应条件下,作者考察了乙烯基氨基甲酸酯的底物范围(Table 2)。各种芳环取代的乙烯基氨基甲酸酯均能很好的适应反应条件,能以良好至优秀的收率和对映选择性得到相应产物。其中,对于乙烯基邻位取代的底物,给电子取代的乙烯基氨基甲酸酯表现出较好的的对映选择性。若R’换成Me,该底物仍然具有较好的耐受性,能以良好的收率和优秀的对映选择性得到相应产物。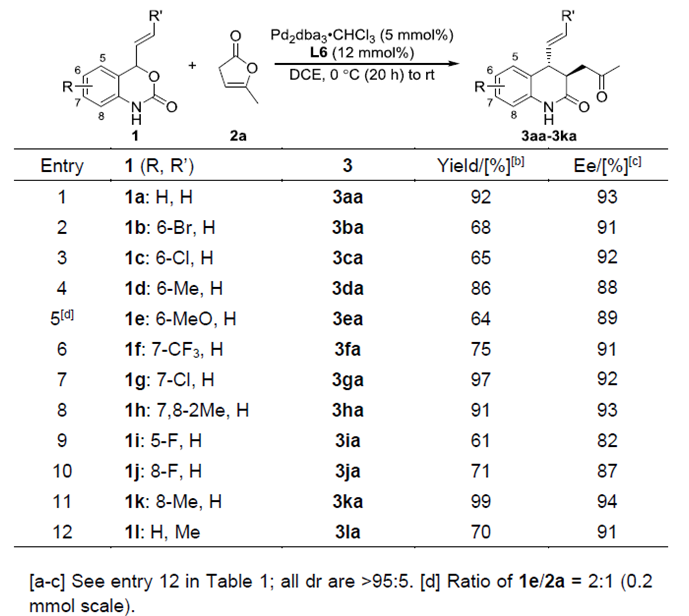
Table 2.乙烯基氨基甲酸酯的底物扩展
紧接着,作者考察了丁烯酸内酯的底物范围(Table 3)。各种苯基取代、苄基取代、萘基取代以及杂环取代的丁烯酸内酯具有较好的耐受性,能以良好至优秀的收率和优秀的对映选择性得到相应产物。α,β-二取代的丁烯酸内酯能较好的适应反应条件,能以良好的收率和对映选择性得到相应产物。而α,γ-二取代的丁烯酸内酯则只能以中等的收率和对映选择性得到相应产物。
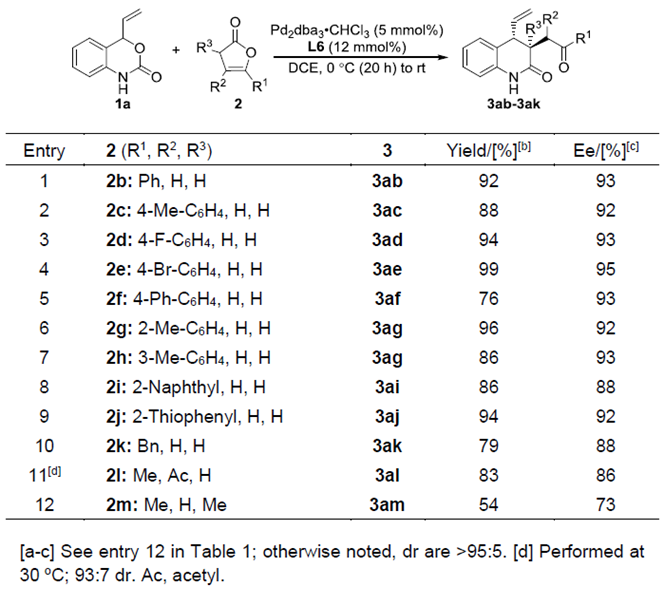
Table 3.丁烯酸内酯的底物扩展
为了更一步了解手性P,S-配体L6的在反应中所起的作用,作者做了密度泛函理论(DFT)计算。根据计算结果,作者四种可能的决定该反应区域选择性和立体选择性的过渡态。其中,TS-Si–Si过渡态的能量最低,所以最稳定。通过分析TS-Si–Si的3D构型,作者发现手性P,S-配体L6的β-萘基对该反应的对映选择性起着至关重要的作用(Scheme 2)。
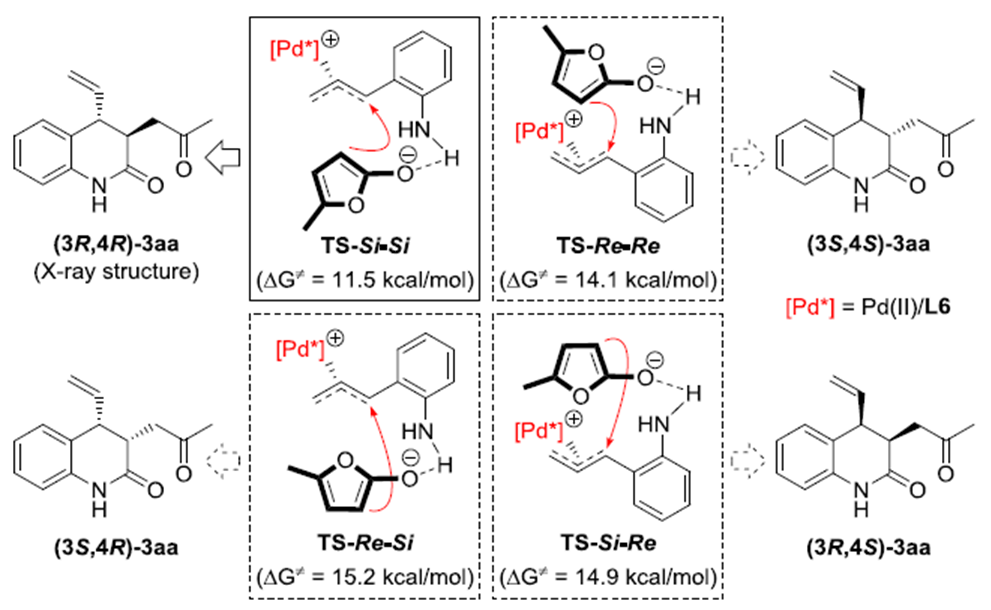
Scheme 2. 立体结构分析
受到上述研究结果的鼓舞,作者对乙烯基氨基甲酸酯和二氢唑酮的反应情况进行了考察(Table 4)。在上述最优条件下,乙烯基氨基甲酸酯和吖内酯也能发生Pd和新型P,S-杂化配体共同催化的不对称[4+2]环加成反应。同时,作者对该反应的底物范围进行了考察(Table 4)。各种C6-C8位取代的乙烯基氨基甲酸酯均能较好的适应反应条件,能以优秀的收率和对映选择性得到相应产物。若将R2的H换成Me,则只能以良好的收率和对映选择性得到相应产物。同时,各种取代的二氢唑酮具有良好的耐受性,能以良好至优秀的收率和对映选择性得到相应产物。

Table 4.乙烯基氨基甲酸酯和二氢唑酮的反应情况
论文总结评价:
华中师范大学陆良秋和肖文精团队报道了第一例Pd和新型P,S-杂化配体催化乙烯基氨基甲酸酯与丁烯酸内酯或二氢唑酮的不对称[4+2]环加成反应,能以高收率和对映选择性得到一系列手性手性二氢喹啉-2-酮化合物。其中,重庆大学的蓝宇教授课题组提供了计算化学支持。
参考文献:
- (a) Wu, B.; Yu, Z.; Gao, X.; Lan, Y.; Zhou, Y.-G. Chem. Int. Ed. 2017, 56, 4006. DOI: 10.1002/anie.201700437 (b) Griswold, J. A.; Horwitz, M. A.; Leiva, L. V.; Johnson, J. S.; J. Org. Chem. 2017, 82, 2276. DOI: 10.1021/acs.joc.6b03059
- Hyde, A. M.; Buchwald, S. L. Lett. 2009, 11, 2663. DOI: 10.1021/ol9007102
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!













No comments yet.