
本文作者:杉杉
导读
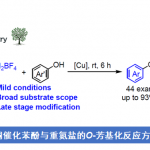
醛与酮类化合物广泛存在于生物质资源 (biomass resource)中,同时,作为重要的合成砌块,在有机合成方法学研究中尤为关键。然而,通过醛或酮的直接去氧偶联过程,实现C(sp3)-C(sp3)键的构建仍具有较多的挑战。近日,加拿大McGill大学李朝军教授课题组Nat. Commun.中发表论文,报道通过醛或酮底物原位形成的腙产物 (空气与湿气稳定)进行的镍催化还原同偶联反应 (reductive homo-coupling)方法学,进而有效地完成C(sp3)-C(sp3)键的构建。这一全新的还原同偶联策略具有底物应用范围广泛、优良的官能团兼容性以及副产物(例如H2O、N2以及H2)环境无害等优势。此外,作者通过上述策略在生物分子合成以及工程塑料 (engineering plastic)聚醚醚酮 (polyetheretherketone, PEEK)模型化合物 (model compound)转化中的应用研究,进一步阐明上述反应策略的合成应用价值。
C(sp3)-C(sp3) bond formation via nickel catalyzed deoxygenative homo-coupling of aldehydes/ketones mediated by hydrazine
D.Cao, C.Li, H. Zeng, Y. Peng, C. Li, Nat. Commun. 2021, 12, 3729. doi: 10.1038/s41467-021-23971-7.
正文
C(sp3)-C(sp3)键的构建,为有机合成转化中最为基本的策略之一。尤其带有C(sp3)-C(sp3)键的联苄基衍生物 (dibenzyl derivative)作为重要的结构单元,广泛存在于天然产物、药物、农用化学品、染料分子以及聚合物分子中。尽管通过二芳基烯以及二芳基炔的还原反应方法学[1]-[2],能够成功实现联苄基化合物的构建。然而,采用同偶联反应策略完成联苄基分子的构建,具有更为显著的优势。例如,近年来,苄基卤[3]、苯基溴化镁[4]、苯乙酸[5]、苯基硼酸[6]以及甲苯衍生物[7]的同偶联反应方法学研究,已取得较多进展。然而,上述方法学中仍存在较多弊端,例如,需要较为繁琐的前官能团化步骤以及采用湿气敏感的金属有机试剂等。
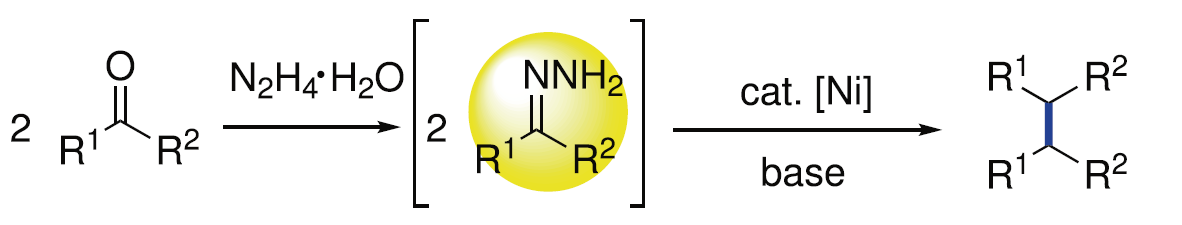
Wolff-Kishner还原反应作为一种经典的直接去氧反应策略[8],能够在肼媒介的条件下,将醛或酮底物转化为一系列烷基衍生物 (Fig. 1a)。受到上述经典反应策略以及本课题组前期研究[9]-[10]的启发,作者设想,能否采用肼作为还原剂,应用于羰基化合物的还原偶联过程,进而实现相应C(sp3)-C(sp3)键的构建。基于上述设想,作者设计出一种在肼媒介条件下,采用镍催化的由醛或酮底物参与的直接去氧同偶联反应方法学,进而成功实现一系列联苄基衍生物的合成 (Fig. 1b)。同时,这一全新的反应策略具有如下优势:(a) 副产物H2O、N2以及H2具有环境无害性;(b) 参与极为易得的醛或酮作为起始原料;(c) 在碱存在的条件下,能够有效避免竞争性副反应过程即Wolff-Kishner还原的发生;(d) 选择更为廉价的镍催化剂;(e) 底物应用范围广泛,并具有优良的官能团兼容性;(f) 能够有效地应用于商品化药物分子的合成以及工程塑料PEEK (polyetheretherketone)相关模型化合物的转化研究。
首先,作者采用通过苯甲醛1a原位形成的腙2a作为模型底物,进行了相关偶联反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用NiCl2作为催化剂,IMeS·HCl作为配体,DBU作为碱,二氧六环作为反应溶剂,反应温度为100 oC,最终以83%收率获得相应同偶联产物3a (entry 25)。

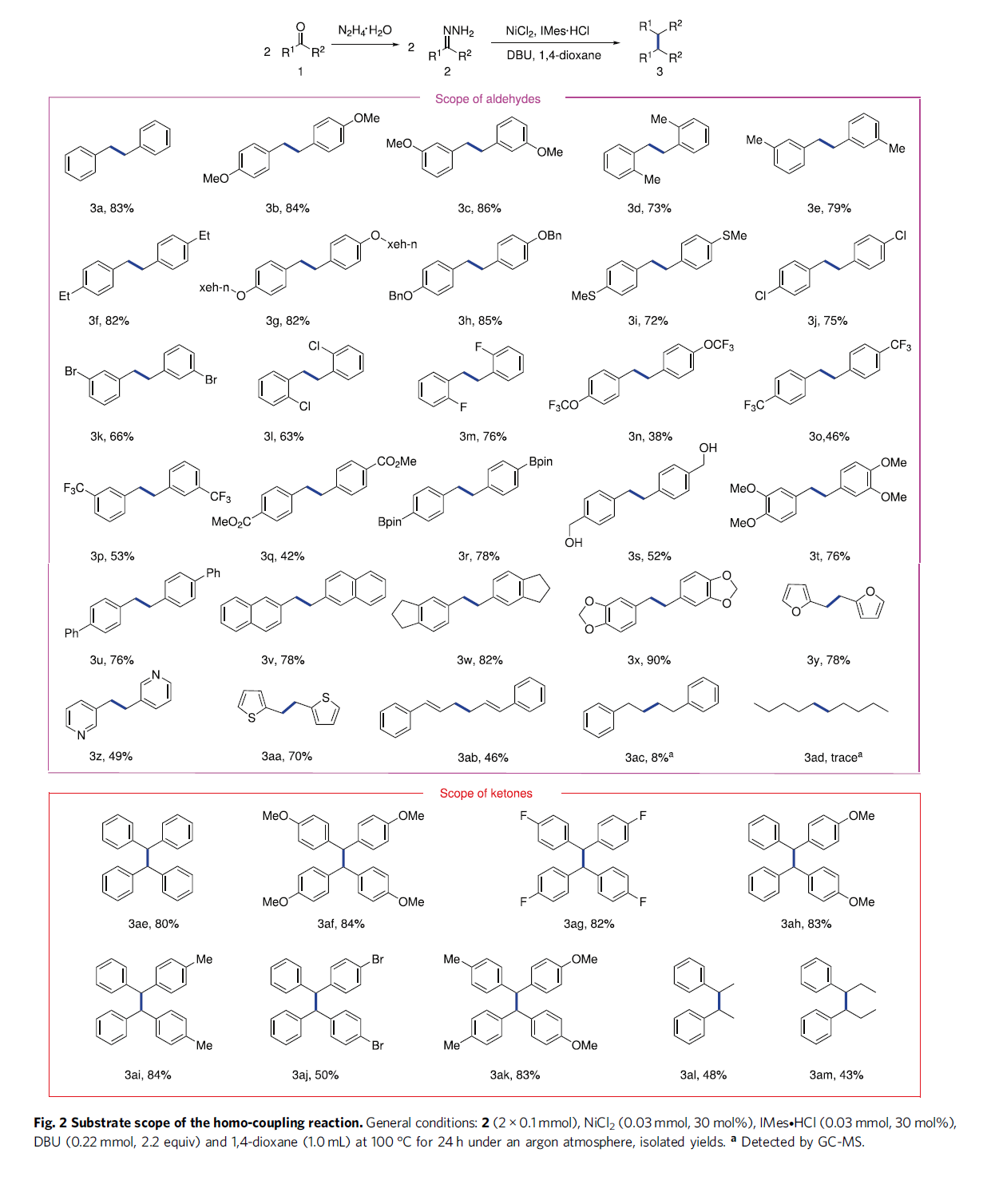
在获得上述最佳反应条件后,作者开始对醛的底物应用范围进行考察 (Fig. 2)。研究发现,一系列芳环不同位置具有吸电子基团与供电子基团取代的芳醛底物,均能够顺利地参与上述的偶联过程,并以38-86%的产率获得相应的同偶联产物3a–3t。同时,作者观察到,各类具有多环 (杂环)芳基取代的芳醛底物,同样能够表现出优良的兼容性,并以中等至优良的收率,获得相应目标产物3u–3aa。之后,该小组发现,对于肉桂醛底物,同样能够以46%反应收率获得相应的1,5-二烯产物3ab。然而,上述标准反应条件对于戊醛与苯乙醛底物,仅能够通过GC-MS检测出8%或痕量收率的产物3ac与3ad,这可能源自于高温条件下,存在形成吖嗪 (azine)产物的竞争性副反应。
接下来,作者进一步对酮的底物应用范围进行考察 (Fig. 2)。研究表明,上述最佳的反应条件对于具有不同类型取代基团的对称二芳基酮底物,均能够良好地兼容,并以优良的收率获得相应目标产物3ae–3ag。同时,作者进一步发现,非对称二芳基酮以及芳基烷基酮底物,同样顺利地参与上述的偶联过程,并获得相应产物3ah–3am,收率为43-84%。
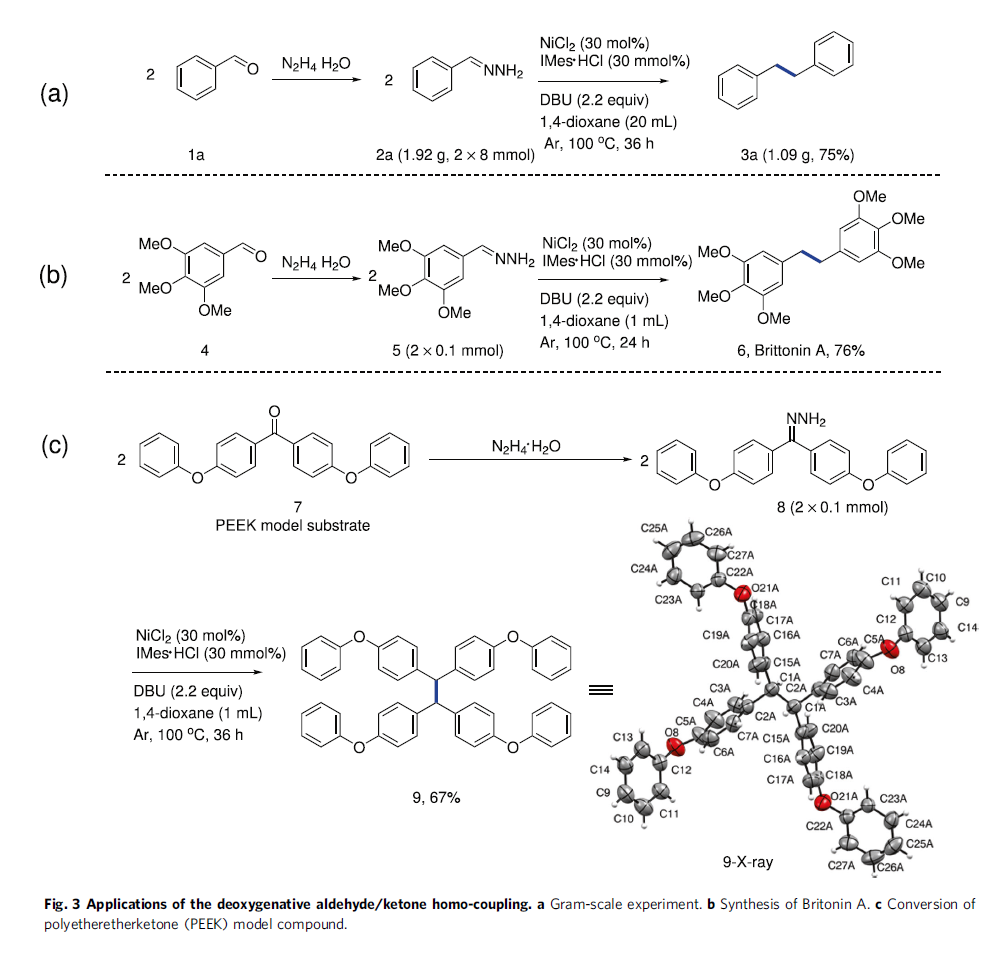
接下来,作者对上述反应策略的合成实用性进行研究 (Fig. 3)。首先,作者在苯甲醛底物1a的克级规模实验研究中发现,上述的标准反应条件下,同样能够获得75%收率 (1.09 g)的目标产物3a (Fig. 3a)。之后,该小组发现,采用3,4,5-三甲氧基苯甲醛作为反应底物,在上述最佳的反应条件下,能够成功完成药物分子Britonin A (6)的构建 (Fig. 3b)。此外,作者进一步发现,工程塑料聚醚醚酮 (PEEK)的模型化合物 (7)在上述镍催化的反应条件下,同样能够有效地转化为相应同偶联产物9,收率为67% (Fig. 3c)。进而表明上述镍催化的还原同偶联策略在复杂分子的后期合成 (late stage synthesis)与修饰研究中具有良好的应用价值。
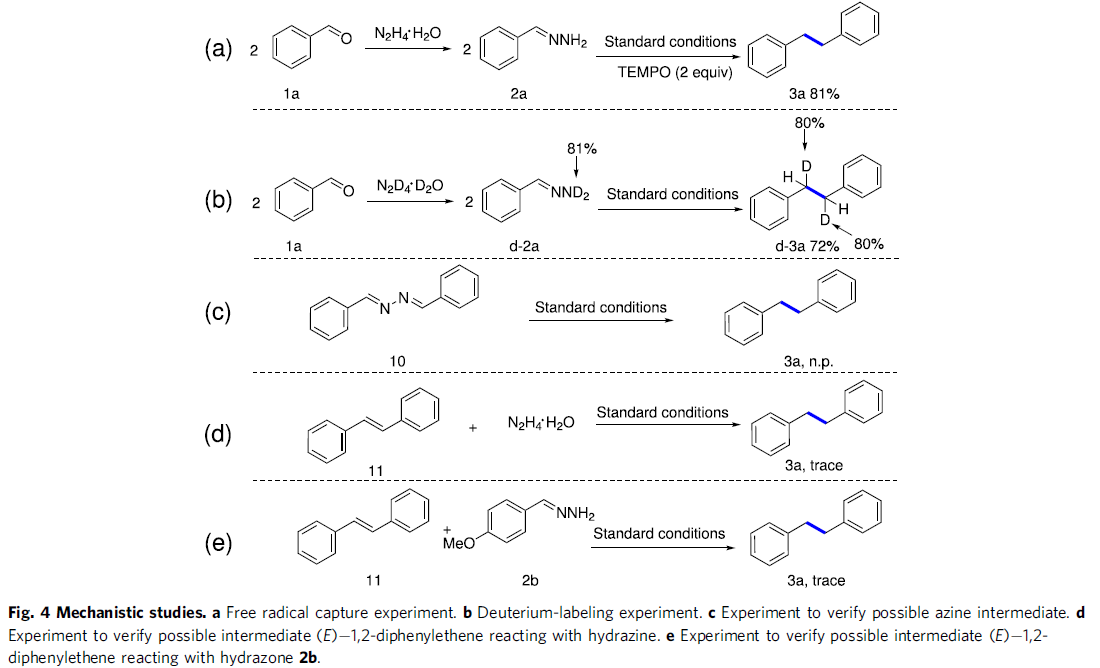
为了阐明合理的反应机理,作者进行一系列相关的控制实验研究 (Fig. 4)。首先,作者发现,在上述标准反应体系中加入TEMPO时,反应未受影响,进而能够排除相应的自由基反应途径 (Fig. 4a)。之后,在采用氘代腙d-2a进行的氘标记实验中,该小组观察到,最终能够获得72%收率的同偶联产物d-3a,其中苄位氘代率为80%,进而表明苄位中的氢源自于腙底物中的N-H基团 (Fig. 4b)。
接下来,作者进一步发现,采用10在上述标准反应条件下参与反应时,未能检测出预期的产物3a,进而表明上述的还原同偶联过程中并未涉及吖嗪 (azine)中间体的参与 (Fig. 4c)。此外,研究表明,在采用(E)-1,2-二苯基乙烯 (11)与水合肼 (Fig. 4d)或腙2b (Fig. 4e)在上述标准条件下进行反应时,仅能够获得较少量的目标产物3a。这一事实表明,(E)-1,2-二苯基乙烯并非上述同偶联过程中的反应中间体。
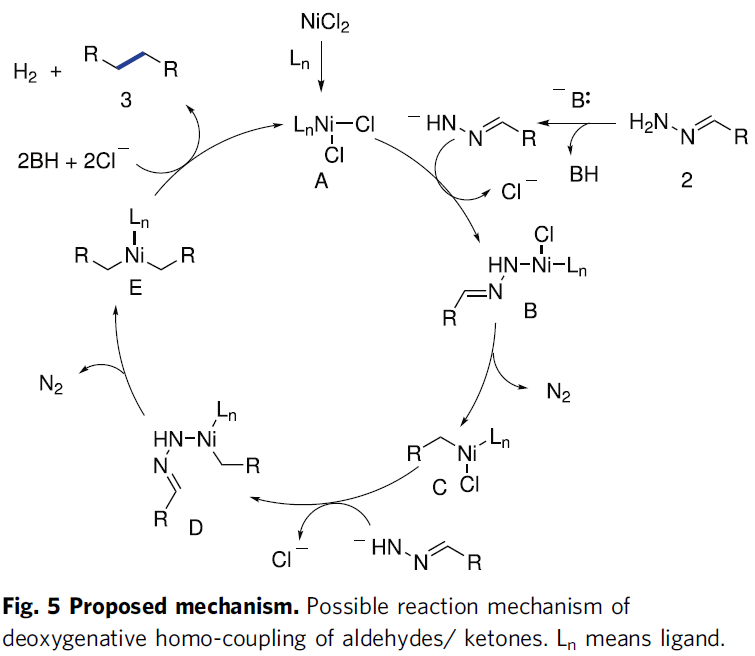
基于上述的实验研究以及前期的文献报道[11]-[12],作者提出一种合理的反应机理 (Fig. 5)。首先,通过反应活性的镍(II)配合物A与腙负离子的配体取代过程,形成镍(II)配合物B,之后,镍(II)配合物B经历类似Wolff-Kishner的反应步骤,释放出N2分子,并通过后续的分子内苄基迁移过程,获得中间体C。接下来,中间体C与另一分子的腙阴离子,再次通过配体取代过程,形成中间体D,之后,D再次经历类似的Wolff-Kishner反应过程以及后续的分子内苄基迁移步骤,释放N2,获得中间体E。最终,通过中间体E的还原消除过程,进而获得相应的同偶联产物3,同时,使活性催化剂A再生。此外,同样可能存在另外一种合理的反应路径,即活性镍配合物A与两分子腙负离子通过配体取代以及后续的分子内迁移、去质子化与N2释放过程,获得预期的还原同偶联产物3。
总结
加拿大McGill大学李朝军教授课题组报道一种通过醛或酮原位形成的腙底物参与的镍催化还原同偶联反应方法学,进而成功完成一系列联苄基衍生物的合成。同时,这一全新的还原同偶联策略具有反应条件温和、底物应用范围广泛、优良的官能团兼容性以及副产物环境无害等优势。此外,该小组通过Brittonin A的合成以及PEEK相关模型化合物的转化研究,进一步阐明上述还原同偶联策略具有良好的合成应用价值。



















No comments yet.