
本文作者:杉杉
导读
与发展较为成熟的中心手性硼化学 (centrally chiral boron chemistry)相比,C-B轴手性化学 (C-B axially chiral chemistry)的发展则具有更多的挑战。近日,华侨大学宋秋玲教授课题组在J. Am. Chem. Soc.中发表论文,首次报道溴代芳烃与非对称双硼试剂 (unsymmetrical diboron reagent)之间的阻转选择性Miyaura硼化反应方法学,进而实现一系列具有光学活性的阻转异构芳基硼化合物的催化合成。同时,该方法学具有底物应用范围广泛、高度的官能团兼容性与良好的反应收率以及优良的对映选择性等优势。
Construction of Axially Chiral Arylborons via Atroposelective Miyaura Borylation
K.Yang, Y.Mao, J. Xu, H. Wang, Y. He, W. Li, Q. Song,
J.Am. Chem. Soc.ASAP. doi: 10.1021/jacs.1c04345.

正文
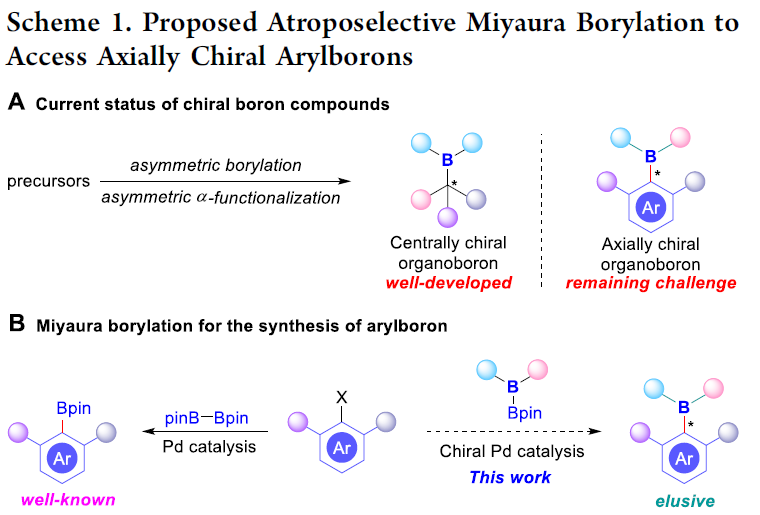
手性有机硼化合物是构建一系列光学活性分子的重要中间体。目前,对于手性有机硼化合物的合成方法学研究,主要涉及不对称硼氢化反应、不饱和键的加成反应、C-H键的硼化反应等。然而,上述的反应策略仅限于具有中心手性有机硼化合物的构建 (Scheme 1A)。与发展较为成熟的中心手性硼化学相比, C-B键轴手性化学的发展仍然较为有限,并具有较大的挑战,这种挑战性可能源自于与较为常见的联芳基化合物 (Csp2–Csp2键长为1.46 Å)相比,较长的Csp2-B (键长为1.58 Å),使旋转能垒偏低,进而易于消旋。并且,由于轴手性骨架广泛存在于手性配体、有机催化剂、天然产物、药物以及材料等分子中,因此,发展一系列有效的阻转选择性控制策略,实现C-B 键轴手性化合物的构建,具有重要意义。同时,作者设想,通过对相关阻转选择性控制策略的深入研究,能够显著提高C-B轴手性化合物骨架的结构多样性,并进一步合成出一系列具有全新特性的有机硼化合物,
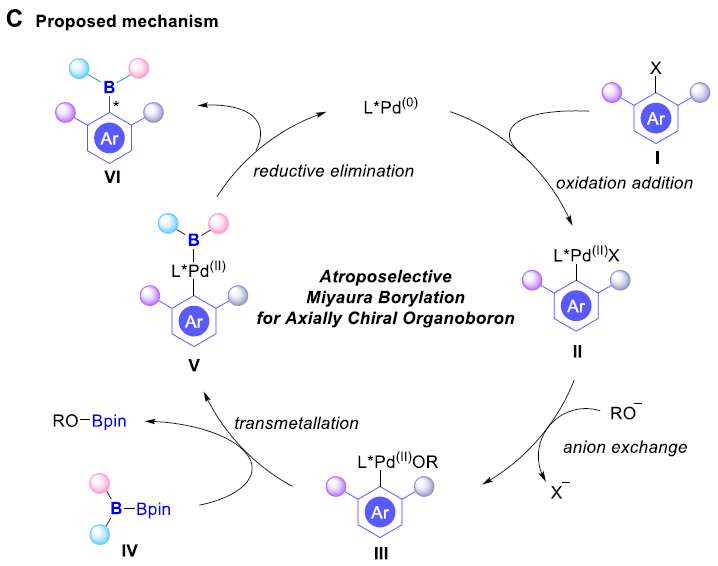
早在1995年,Miyaura课题组[1]率先报道采用钯催化剂参与的芳卤与B2pin2之间的偶联反应方法学,从而顺利实现各类芳基硼化物的构建,并且,文献中将这一反应方法学命名为“Miyaura硼化反应” (Scheme 1B, left)。基于上述研究,华侨大学宋秋玲教授课题组首次报道采用芳卤与非对称双硼试剂 (unsymmetrical diboron reagent)之间的阻转选择性Miyaura硼基化反应,并有效地完成一系列轴手性有机硼化物的直接催化合成 (Scheme 1B, right)。同时,作者提出一种较为合理的反应机理 (Scheme 1C)。首先,芳卤I与光学活性的Pd(0)配合物之间通过氧化加成步骤,形成手性芳基钯中间体II。之后,通过芳基钯中间体II与碱之间的阴离子交换过程,形成中间体III,接下来,III再与非对称双硼试剂IV经转金属化过程,形成中间体V。最终,中间体V经历还原消除步骤,即可获得轴手性芳基硼化合物VI,并使Pd(0)催化剂再生,进而完成相应的催化循环过程。
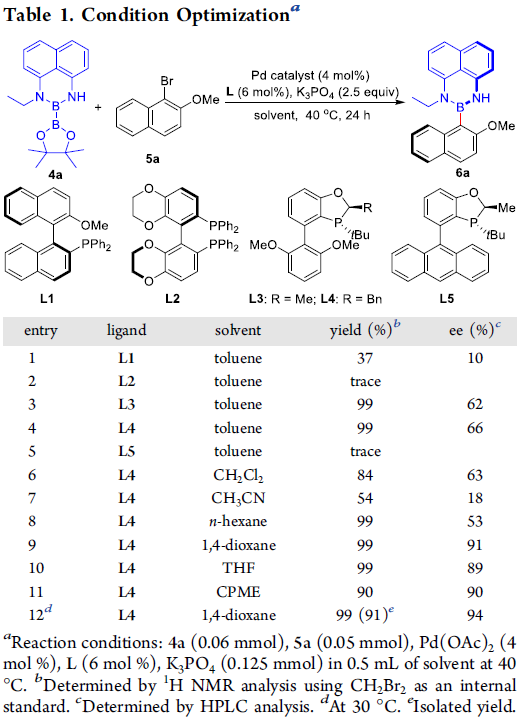
值得注意的是,非对称双硼试剂的选择,对于阻转选择性Miyaura硼基化反应的成功进行至关重要。通常,非对称双硼试剂的选择,需要满足如下要求: (1) 硼原子中具有两种不同类型的取代基[2] (2)能够在反应过程中选择性地进行相应的硼化反应。对于现有的非对称双硼试剂pinB-Bdan (dan:1,8-萘二胺),能够实现有机卤代物与不饱和化合物之间的选择性硼化反应,并获得R-Bdan产物。因此,作者设想,采用氮原子中具有离子不同取代基的pinB-Bdan(R1R2) 试剂,则可能获得相应的轴手性芳基硼化合物。因此,作者开始进行一系列非对称双硼试剂4的制备研究 (Scheme 2)。

在成功完成一系列非对称双硼试剂4的制备之后,作者首先选择非对称双硼试剂4a与1-溴-2-甲氧基萘5a作为模型底物,进行相关反应条件,例如配体、溶剂等的优化筛选 (Table 1)。确定最佳的反应条件为:采用4 mol%的Pd(OAc)2作为催化剂,6 mol%的L4作为配体,2.5 eq.的K3PO4作为碱,二氧六环作为反应溶剂,反应温度为30 oC,最终获得91%收率与94%ee的轴手性硼化合物6a。
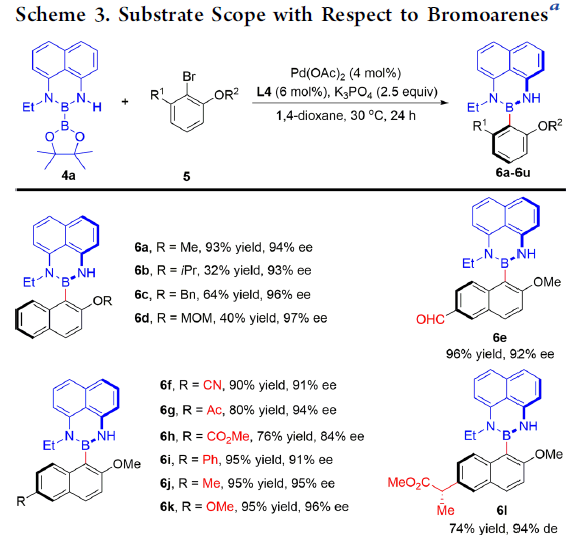
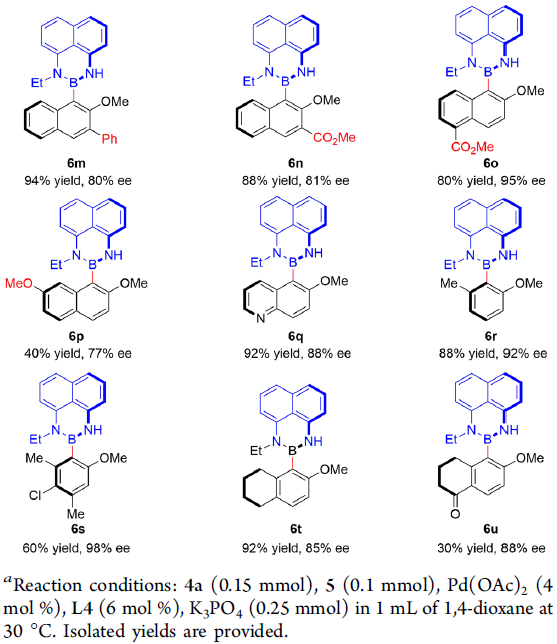
在获得上述最佳反应条件后,作者开始对芳基溴 5的底物适用范围进行考察(Scheme 3)。研究表明,在1-溴萘底物中OR2为不同的取代基时,对于产物对映选择性的无显著影响 (6a–6d),然而,却因立体位阻的存在,使反应收率有所降低 (6b)。同时,作者发现,1-溴萘底物中R1为3-、5-或6-位取代时,均能够顺利与4a反应,并获得相应的产物 6e–6o。然而, 1-溴萘中R1为7-甲氧基取代时,则不利于硼化反应的进行,仅能够获得40%收率与77% ee的产物 6p。更为重要的是,这一全新的阻转选择性Miyaura硼化反应方法学具有温和的反应条件与良好的官能团兼容性,例如醛基、氰基、酮基、酯基等均能够与上述反应的最佳条件良好地兼容。并且,上述的硼化反应条件同样能够应用于溴代喹啉衍生物 5q,并获得的92% 收率88% ee的产物 6q。 此外,上述的硼化方法学同样能够顺利应用于具有光学活性的phenyl-Bdans (6r–6u) 的阻转选择性合成 (85-98% ee)。
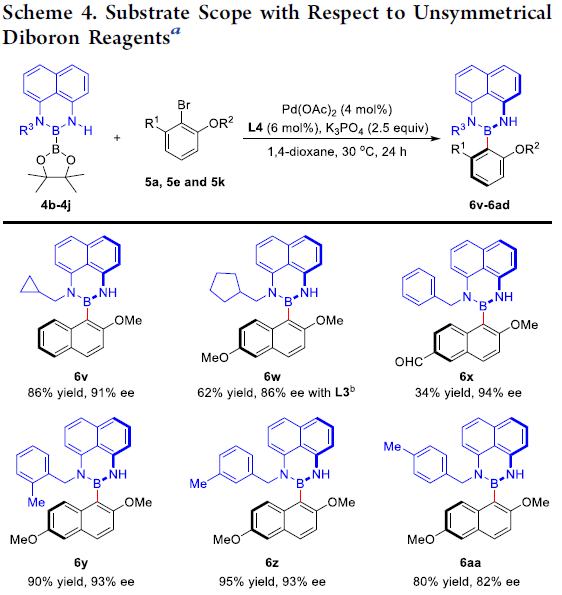
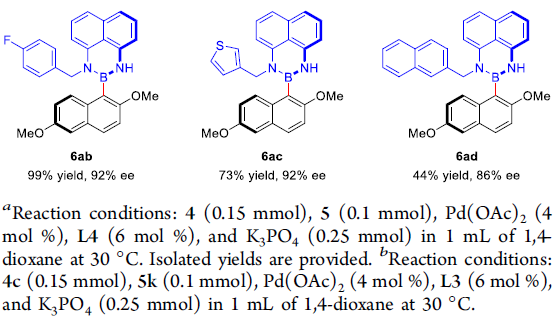
接下来,作者进一步对非对称双硼试剂的应用范围进行研究 (Scheme 4)。实验发现,双硼试剂中的R3取代基为环丙甲基、环戊甲基、苄基、噻吩-2-甲基以及萘-2-甲基时,均能够顺利地参与上述的阻转选择性硼化过程,并获得相应轴手性产物6v–6z与6aa–6ad,收率为34-99%,ee为82-94%。
同时,作者对上述硼化反应方法学的合成应用价值进行深入研究 (Scheme 5)。首先,作者发现,将4a与5a在标准条件下,进行相应克级规模实验时,同样能够获得95%收率与94%ee的轴手性芳基硼化合物6a。并且,轴手性芳基硼化合物6a能够与n-BuLi反应,形成氨基锂中间体,并进一步与一系列不同类型的亲电试剂反应,最终获得一系列相关的衍生化产物7a–7d,并且,反应结束后,产物7a–7d的对映纯度保持不变。此外,之前已有文献报道,手性膦配体产物7c与7d能够成功应用于Pd催化的不对称烯丙基化反应方法学研究,从而进一步表明衍生化产物7c与7d作为新型膦配体,具有潜在的合成应用价值。

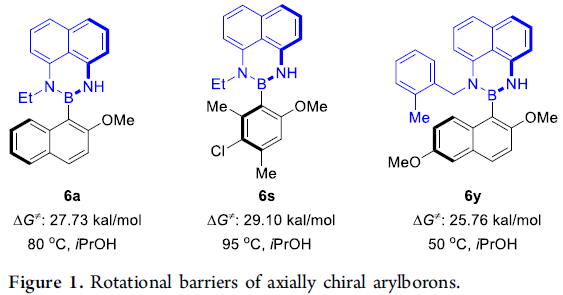
为进一步阐明相应轴手性芳基硼化合物的立体化学稳定性,作者进行相关的外消旋化实验 (racemization experiment)研究 (Figure 1)。首先,作者发现,与6a(27.73 kal/mol)以及6s(29.10 kal/mol)的旋转能垒进行相比,轴手性苯基-Bdan比萘基-Bdan具有更高的稳定性,这可能源自于甲基比萘中的methide具有更高的立体位阻。接下来,作者进一步观察到,采用带有较小取代基的底物6y时,围绕C-B轴的立体位阻进一步减弱,因此,旋转能垒较低 (25.76 kal/mol)。上述结果表明,C-B轴手性化合物的合成需克服因低旋转能垒的存在,而伴随的易产生外消旋化的问题。此外,实验过程中,该小组进一步发现,邻位四取代的轴手性芳基硼化合物7a同样具有良好的构型稳定性,即使在150℃下,放置24 h,仍未观察到相应外消旋化过程的发生。
总结
宋秋玲教授课题组报道首例采用钯催化的溴代芳烃与非对称双硼试剂之间的阻转选择性Miyaura硼化反应方法学,进而高效地完成一系列轴手性芳基硼化合物的构建。同时,这一全新的阻转选择性Miyaura硼化反应方法学具有底物适用范围广泛、较高的反应收率与良好的官能团兼容性以及优良的对映选择性等优势。此外,通过克级规模实验以及后期的衍生化反应研究,进一步阐明该方法学在有机合成中的应用价值。
参考文献
[1] (a) T. Ishiyama, N. Matsuda, N. Miyaura, A. Suzuki, J. Am. Chem. Soc. 1993, 115, 11018. doi: 10.1021/ja00076a081.(b) T. Ishiyama, M. Murata, N. P. Miyaura, J. Org. Chem. 1995, 60, 7508. doi: 10.1021/jo00128a024
[2] G. Bringmann, A. J. P. Mortimer, P. A. Keller, Gresser, M. J. Garner, J. Breuning, M. Angew. Chem. Int. Ed. 2005, 44, 5384. doi: 10.1002/anie.200462661.
















No comments yet.