

本文作者:杉杉
导读
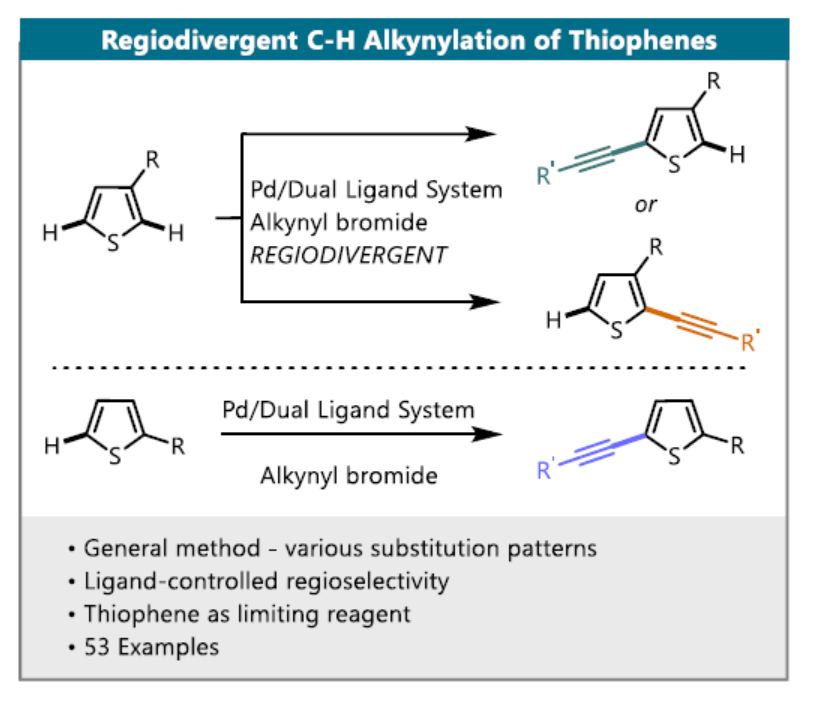
炔基结构广泛存在于天然产物和生物活性分子中,同时也可作为多功能性的基团用于后期的衍生化。常规的合成方法为通过卤化及Sonogashira交叉偶联,将炔烃引入杂芳烃中,该方法的区域选择性完全取决于卤化步骤。类似地方法也可实现噻吩的直接炔基化。近日,德国明斯特大学(Westfälische Wilhelms-Universität Münster)Manuel van Gemmeren课题组在Angew. Chem. Int. Ed.上发表论文,报道了钯催化噻吩的C-H活化/炔基化反应。解决了噻吩C3取代基的区域选择性(C2或C5炔基化)难题,通过两组反应条件实现区域发散反应,从而选择性地合成目标异构体。此外,该方法具有广泛的底物范围,为后期修饰提供了多种可能。
Catalyst Controlled Regiodivergent C-H Alkynylation ofThiophenes
Arup Mondal, and Manuel van Gemmeren*
Angew. Chem. Int. Ed. ASAP DOI:10.1002/anie.202012103 https://doi.org/10.1002/ange.202012103
正文
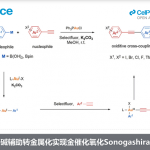
噻吩结构广泛存在于材料科学和药物化学中,噻吩的直接功能化是获取具有价值的噻吩衍生物的重要手段之一。炔基作为有机化学中常见的多功能性官能团,将其插入(杂)芳烃中最常用的方法是Sonogashira交叉偶联,其中产物形成的区域选择性取决于卤化步骤。鉴于炔基化噻吩在药物和有机材料中的重要性,迫切需要一种可直接获取这些产物的替代方法(具有原子经济性),并且在区域选择性面临挑战的情况下,也可以提供替补的产物。2010年,Waser[1]课题组报道了通过金和Brønsted酸催化实现的2-位取代噻吩的C5炔基化反应,同时获得一例3-位上含有富电子的C2炔基化的产物(Scheme 1)。此外,Su[2]课题组报道了Pd催化的2-取代的噻吩和苯乙炔的氧化交叉偶联。然而,目前尚未获得实现3-位取代噻吩的选择性炔基化的通用方法。
对于3-位取代的噻吩,C-H活化的区域选择性主要由取代基的空间和电子性质以及催化剂体系的敏感性决定。对于3-位上带有供电子取代基(EDG)的底物,通常获得C2产物。相反,对位阻敏感的催化剂为了避免和3-位取代基之间的空间冲突,从而形成C5产物。对于3-位上具有吸电子基团(EWG)时(具有路易斯碱性),可作为导向基团(DG),通过螯合控制促进C2位官能团化。2017年,Zeng[3]课题组报道了一种Ir催化芳烃邻位的直接炔基化反应,其中包括一例噻吩底物(3-位取代有羧酸酯导向基团)的C2炔基化的例子(Scheme 1)。同样,在2017年,Echavarren[4]课题组报道了Ru催化的芳烃邻位炔基化反应,并获得3-取代噻吩的C2和C4位双炔基化产物,同时还报道了一例使用4-溴噻吩-3-羧酸作为底物,获得C2单炔基化产物的工作(Scheme 1)。在2018年,同一课题组[5]报道了Rh催化的芳烃邻位炔基化反应,其中包括一个3-取代噻吩与苄基醚作为弱DG来获得C2产物的例子。综上所述,噻吩的直接C-H炔基化仍然是一个极具挑战性但具吸引力的课题。迄今为止,更具挑战性的3-取代噻吩底物选择性官能化仍是一项难题。
基于上述的结果以及该课题组在控制杂芳烃C-H活化区域选择性方面的经验,作者认为,通过合理催化剂的设计,可实现噻吩C2和C5选择性炔基化。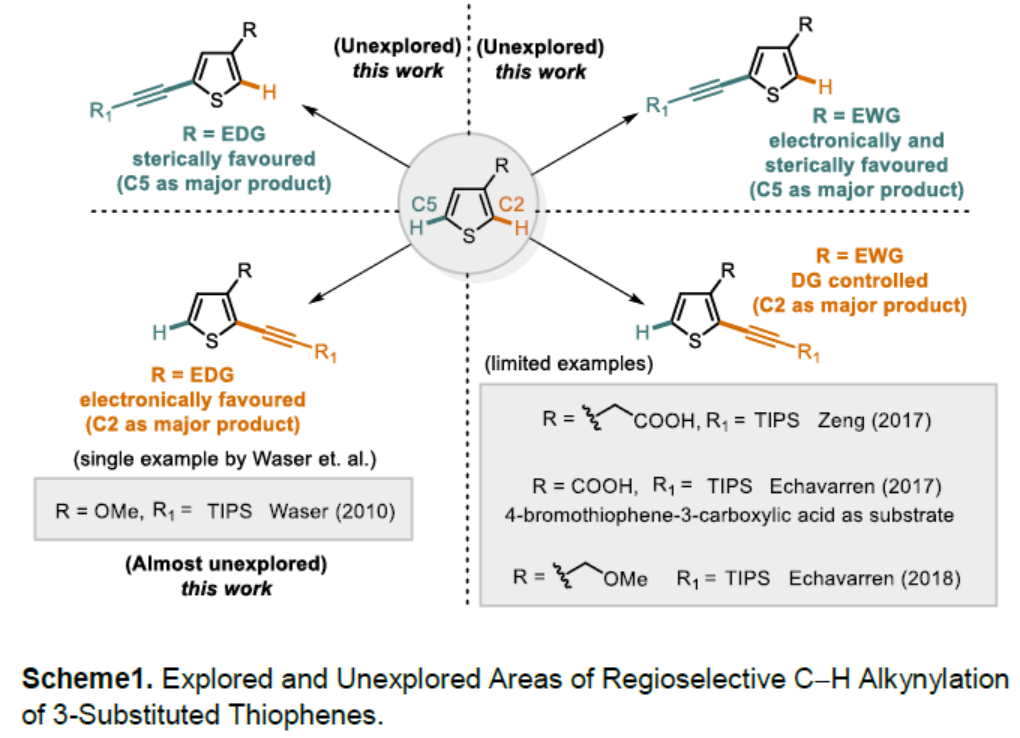
首先,作者以3-己基噻吩1a和溴代炔烃2a作为模型底物,进行了相关炔基化反应条件的筛选(Scheme 2)。通过对配体的设计,作者发现,使用L4时,以高收率(71%)和区域选择性(94∶6)获得目标化合物3a-C5。此外,对照实验表明(Table 1),该反应需具有双重配体,缺少任何一种配体时反应结果明显较差。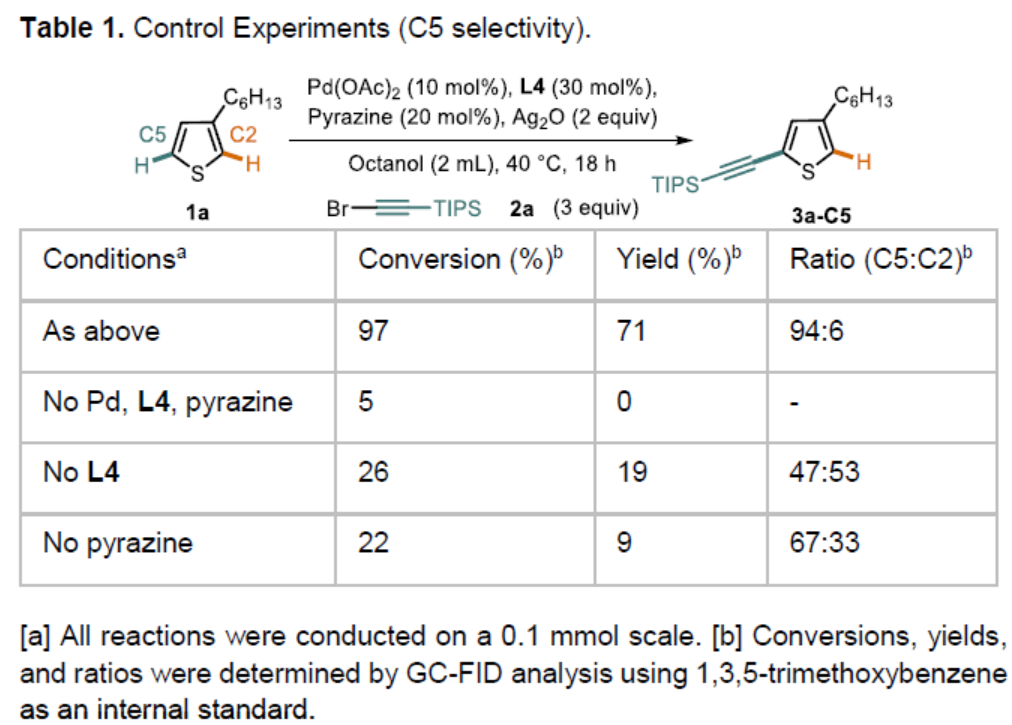
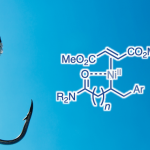
在获得上述最佳反应条件后,作者开始对噻吩底物1进行了扩展(Scheme 3)。通过产物3a-C5至3c-C5的对比发现,烷基取代基的空间位阻降低会使C5选择性降低。具有酯、醚、酰基取代基的底物,也具有良好的C5选择性(3d-C5至3h-C5)。3-芳基取代的噻吩的底物,芳基不受电子效应影响,均可获得相应的产物3i-C5至3m-C5。此外,非天然氨基酸衍生物(3n-C5)和雌酮衍生物(3o-C5)也成功地进行了炔基化,进一步体现了反应的实用性。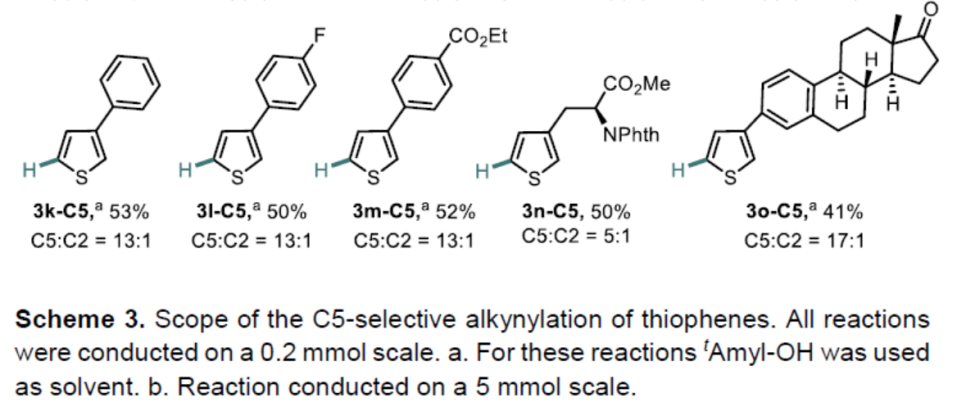
随后,作者开始开发C2选择性催化剂体系,作者认为通过提高催化剂的亲电性,可以实现电子控制(Scheme 4)。当配体上具有更强的吸电子取代基L8时,可获得4:1的选择性,有利于C2产物的生成。此外,作者还对其它参数进行了优化,以进一步提高收率和选择性。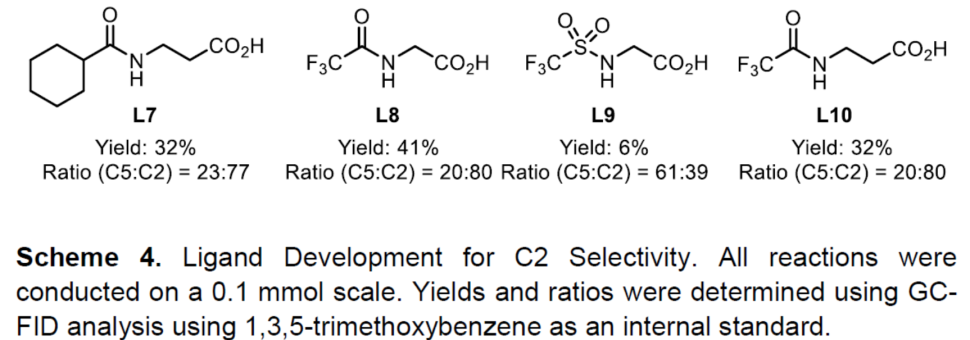
紧接着,作者对C2炔基化的范围进行了扩展(Scheme 5)。当烷烃的取代基的位阻越小,则C2选择性越好(3a-C2-3c-C2)。值得注意的是,含有卤代基(3p-C2和3q-C2)以及强供电子的甲氧基(3r-C2)基团时,仅产生C2产物。同时,各类芳基取代的噻吩,均可实现C2炔基化反应,生成产物3i-C2至3m-C2。值得注意的是,苯环上的供电子甲氧基取代基(3i-C2)比缺电子酯取代基(3m-C2)具有更高的C2选择性。同时,该催化剂体系也成功的对非天然氨基酸衍生物(3n-C2)和雌酮衍生物(3o-C2)的C2炔基化反应。
此外,作者对溴代炔烃2的底物范围也进行了扩展(Scheme 6)。反应结果表明,在标准的C2和C5炔基化的条件下,均可获得令人满意的产率和相应的选择性,从而获得4-C5至12-C5和4-C2至12-C2。然而,具有α-非季取代基(如环己基或正己基)的溴代炔烃以及苯乙炔衍生的溴代炔烃均未形成任何产物,但可先进行上述标准反应后,再除去TIPS-基团,然后进行Sonogashira交叉偶联,即可获得相应的目标产物。
同时,对于区域选择性具有较低挑战性的2-取代基噻吩(如乙基、甲氧基、苯、卤素),同样可在上述的标准条件下反应,获得相应的目标产物14a-14e。同样,2,3-二取代(14f)也具有良好的耐受性。因此,该催化体系可作为噻吩炔基化的通用方法(Scheme 7)。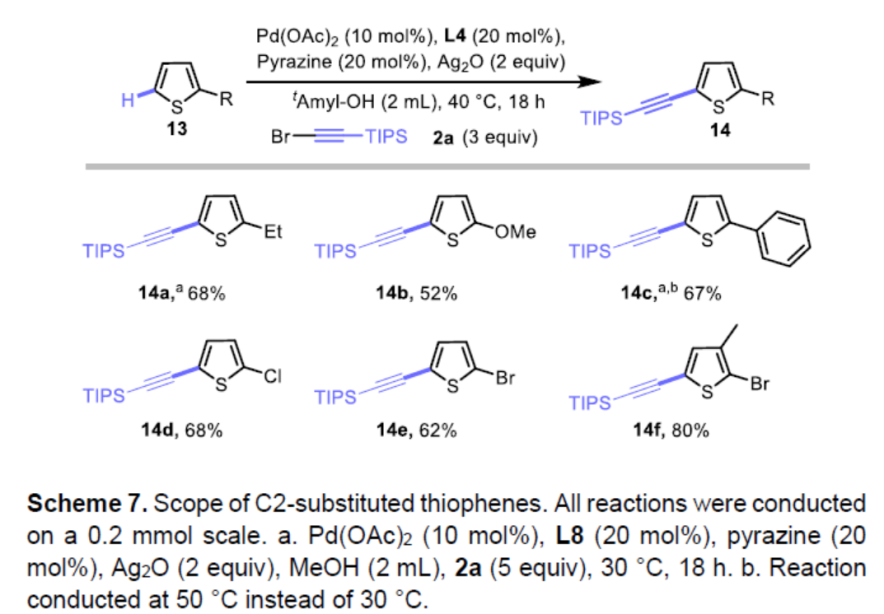
总结
德国明斯特大学Manuel van Gemmeren课题组报道了利用一对通用Pd催化体系,分别实现具有挑战性3-取代噻吩底物的C5-和C2-炔基化反应,获得相应的C5-和C2-炔基化产物的工作。同时,该催化剂体系也成功的对非天然氨基酸衍生物和雌酮衍生物进行了相关的炔基化反应,进一步体现了反应的实用性。
参考文献
[1] J. P. Brand, J. Waser, Angew. Chem. Int. Ed. 2010, 49, 7304.[2] a) X. Jie, Y. Shang, P. Hu, W. Su, Angew. Chem. Int. Ed. 2013, 52,3630; b) additionally, a single example of a C5-selective alkynylationwith a 2,3-disubstituted thiophene substrate has been described by Liand coworkers: B. Liu, W. Ouyang, J. Nie, Y. Gao, K. Feng, Y. Huo, Q.Chen, X. Li, Chem. Commun2020, DOI: 10.1039/D0CC04739B.
[3] C. Chen, P. Liu, J. Tang, G. Deng, X. Zeng, Org. Lett. 2017, 19, 2474.
[4] E. Tan, A. I. Konovalov, G. A. Fernández, R. Dorel, A. M. Echavarren,Org. Lett. 2017, 19, 5561.
[5] E. Tan, O. Quinonero, M. Elena de Orbe, A. M. Echavarren, ACS Catal.2018, 8, 2166.


















No comments yet.