
本文作者:杉杉
导读
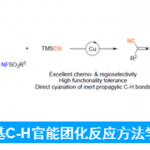
近日,美国Rutgers大学的M. Szostak课题组在Org. Lett.中发表论文,报道采用钯催化剂参与的羧酸脱羰Sonogashira交叉偶联反应 (decarbonylative Sonogashira cross-coupling)方法学。该方法学通过羧酸的原位活化,形成混合酸酐,并采用Pd(OAc)2/Xantphos体系,进行后续的脱羰过程,形成芳基-钯中间体,继而与一系列炔基化合物进行相应的偶联反应。并且,反应过程中涉及通过羧酸底物参与的Pd(0)/ Pd (II)催化循环过程。同时,通过这一全新的脱羰Sonogashira交叉偶联策略,能够有效地完成C(sp2)-C(sp)键的构建,并进一步应用于药物分子的后期衍生化。此外,反应机理研究表明,在转金属化步骤之前,首先需要进行羧酸底物的脱羰过程。
Decarbonylative Sonogashira Cross-Coupling of Carboxylic Acids
C. Liu, M. Szostak, Org. Lett. ASAP. doi: 10.1021/acs.orglett.1c01445.

正文
Sonogashira交叉偶联反应,是将炔基直接引入有机分子的一种应用最为广泛并高效的反应策略。而且,Sonogashira交叉偶联反应同样能够良好地应用于相关的工业生产过程。其中,炔基化合物作为有机合成中的关键砌块,能够完成一系列重要的合成转化过程。在Sonogashira交叉偶联方法学中,典型的亲电底物主要涉及芳卤与芳基拟卤代物,例如芳基磺酸酯。近年来,碘鎓盐、重氮盐、鏻盐以及锍盐等其他亲电底物参与的Sonogashira交叉偶联方法学,已经受到有机合成化学家的广泛关注,进而能够实现相应C-X、C-O、C-N、C-P以及C-S键的断裂[1]。在上述转化过程中,大多采用Pd/Cu共催化体系[2],然而,对于仅选择钯催化剂参与的Sonogashira交叉偶联方法学,则较少有文献报道[3]。此外,近期已经成功设计出通过导向基团辅助的C-H炔基化策略[4]。
之前,本课题组已经成功开发出一系列采用羧酸底物参与的脱羰交叉偶联反应方法学[5]。在上述策略中,通过羧酸的原位活化,形成混合酸酐,之后,通过低价金属试剂对C(O)-OR键的选择性氧化加成以及后续的脱羰过程[6],形成芳基-金属中间体,再经历还原消除过程,即可获得相应的偶联产物 (Figure 1)。受到上述研究报道的启发,这里,作者成功设计出一种采用钯催化剂参与的羧酸脱羰Sonogashira交叉偶联反应方法学策略。
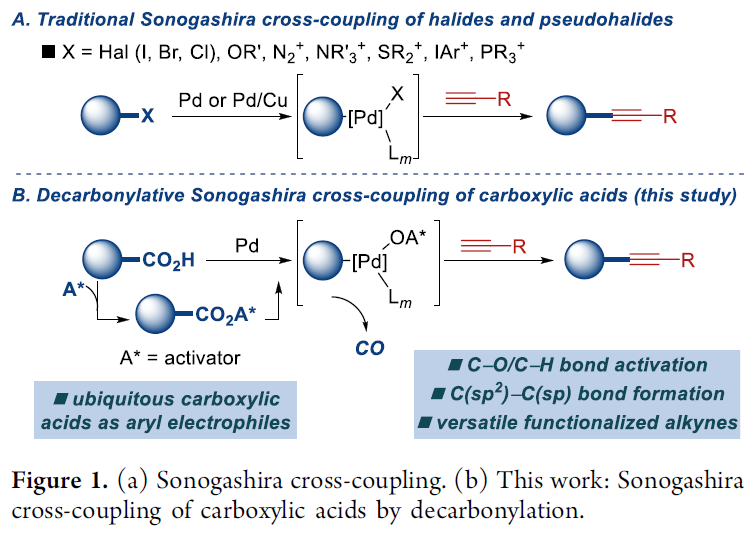
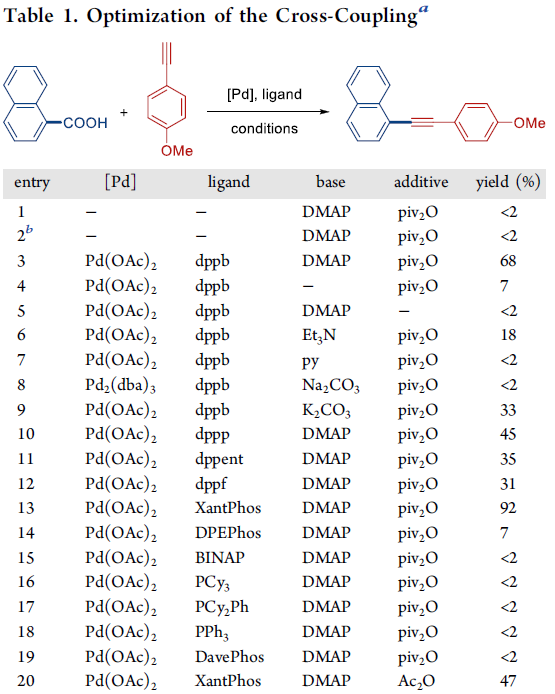
首先,作者采用萘甲酸与4-甲氧基苯乙炔作为模型底物,进行相关偶联反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:采用5 mol% Pd(OAc)2作为催化剂,10 mol% Xantphos作为配体,DMAP作为碱,同时加入piv2O作为添加剂,二氧六环作为反应溶剂,反应温度为160 oC,最终获得92%收率的偶联产物。

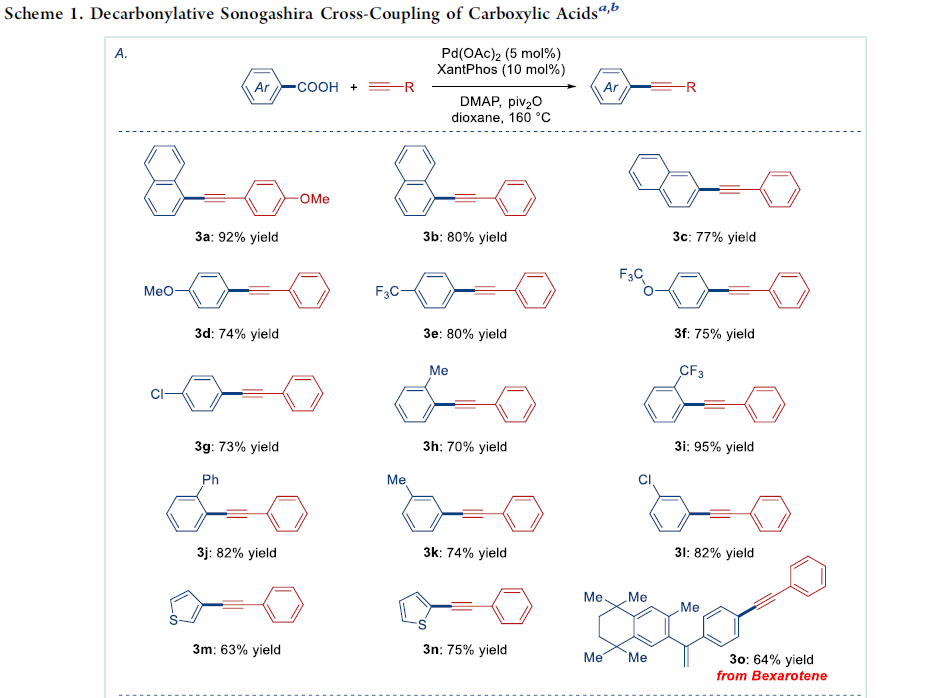
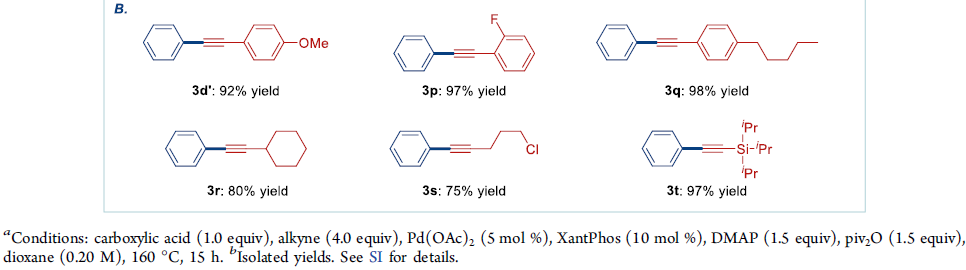
在获得上述的最佳反应条件之后,作者分别对羧酸与炔基化合物的适用范围进行考察 (Scheme 1)。首先,作者发现,不同取代位置的萘甲酸底物,均能够顺利参与上述的偶联过程,并获得产物3a–3c,收率为77-92%。同时,芳环中具有吸电子与供电子基团取代的苯甲酸底物,均能够与上述标准反应条件良好地兼容,并获得相应偶联产物3d–3l,收率为70-95%。并且,杂环羧酸 (例如噻吩甲酸)底物,同样能够较好地参与上述的交叉偶联过程,并获得产物3m与3n。
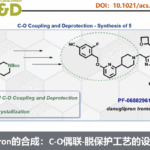
值得注意的是,抗肿瘤剂 (antineoplastic agent) Bexarotene在上述的标准条件下,同样能够较好地完成相应的脱羰Sonogashira交叉偶联反应,并获得衍生化产物3o,收率为64%。产物,作者发现,这一氧化还原中性交叉偶联策略中的脱羰步骤,对于底物中取代基立体与电子效应的影响较不敏感。因此,这一全新的脱羰Sonogashira交叉偶联策略,已经成为向有机分子中引入炔基的一种更为通用的反应策略。
接下来,作者进一步对炔基底物是适用范围进行研究。该小组发现,芳环中具有不同基团取代的苯乙炔底物,均能够顺利地参与上述的偶联反应过程,并获得产物3d’、3p与3q,收率为92-98%。并且,上述的最佳反应条件对于烷基乙炔底物 (例如环己基乙炔),同样能够以80%的收率,获得相应偶联产物3r。值得注意的是,上述的最佳反应条件,对于带有敏感性烷基亲电基团 (例如卤烷基)取代的芳炔底物,同样能够良好地进行兼容,并获得产物3s,收率为75%,同时,偶联产物3s同样能够进行进一步的官能团化。此外,作者发现,硅基乙炔底物 (例如(三异丙基硅基)乙炔),同样能够顺利地完成相应的偶联反应,并获得产物3t,收率为97%,同时,3t通过后续的去保护步骤,能够进一步获得相应的端炔产物。
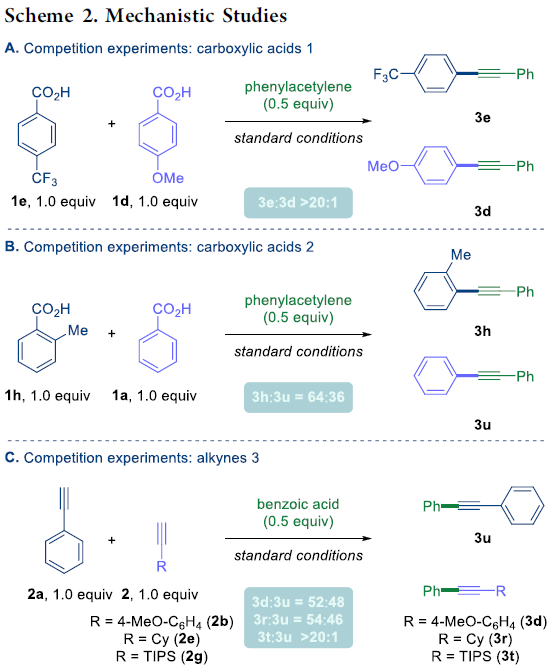
之后,作者对上述偶联过程的反应机理进行深入研究 (Scheme 2)。首先,作者选择具有不同取代基团的苯甲酸底物 (1e与1d)进行相关的分子间竞争实验,进而观察到,芳环中带有吸电子基团取代的苯甲酸底物具有更高的反应活性,其中3e:3d的产物比> 20:1 (Scheme 2A)。之后,作者发现,具有较高立体位阻的苯甲酸底物,则具有较高的反应活性,这与通过酰基钯配合物立体效应,进而有利于脱羰过程[6]进行的实验事实相一致 (Scheme 2B)。相反地,作者在选择有不同基团取代的乙炔底物,进行的分子间竞争实验中观察到,富电子基团取代的的乙炔底物具有更高的反应活性 (Scheme 2C)。
接下来,该小组发现,将混合酸酐4与炔基化合物2b在上述的标准条件下进行反应,能够获得82%收率的脱羰交叉偶联产物3a (Scheme 3A)。同时,作者进一步考察炔酮化合物5与6潜在的脱羰性能,然而,实验中作者发现,在上述的标准条件下,仅能够分别获得极为少量的目标产物3b与2h (Scheme 3B)。此外,作者同样发现,苯乙炔底物在上述的标准反应条件下,能够获得29%收率的二聚产物二苯乙炔。综上结果表明,反应机理路径中,在转金属化步骤之前,首先需要完成羧酸底物的脱羰过程。

总结
美国Rutgers大学M. Szostak课题组报道一种钯催化的采用羧酸底物参与的脱Sonogashira交叉偶联反应方法学。在这一全新的偶联策略中,选择Pd(OAc)2/Xantphos催化体系,其中,反应过程涉及混合酸酐的选择性氧化加成以及后续的脱羰步骤。并且,上述的偶联反应策略能够良好地与一系列羧酸以及炔基底物进行兼容,进而较为容易地完成C(sp2)-C(sp)键的构建。此外,这一偶联策略同样能够有效地应用于药物分子的衍生化。最后,通过反应机理的研究表明,上述的交叉偶联过程中涉及脱羰、转金属化等步骤。
参考文献
[1] (a) L. Hwang, Y. Na, J. Lee, Y. Do, S. Chang, Angew. Chem. Int. Ed. 2005, 44, 6166. doi: 10.1002/anie.200501582.(b) G. Fabrizi, A. Goggiamani, A. Sferrazza, S. Cacchi, Angew. Chem. Int. Ed. 2010, 49, 4067. doi: 10.1002/anie.201000472.
(c) D. Zhu, Y. Wu, B. Wu, B. Luo, A. Ganesan, F. Wu, R. Pi, P. Huang, S. Wen, Org. Lett. 2014, 16, 2350. doi: 10.1021/ol5006714.
(d) Z. Tian, S. Wang, S. Jia, H. Song, C. Zhang, Org. Lett. 2017, 19, 5454. doi: 10.1021/acs.orglett.7b02764.
[2] M. Eckhardt, G. Fu, J. Am. Chem. Soc. 2003, 125, 13642. doi: .doi: 10.1021/ja038177r.
[3] X. Pu, H. Li, T. J. Colacot, J. Org. Chem. 2013, 78, 568. doi: 10.1021/jo302195y.(b) M. Aufiero, F. Proutiere, F. Schoenebeck, Angew. Chem. Int. Ed. 2012, 51, 7226. doi: 10.1002/anie.201202504.
(c) M. Gazvoda, M. Virant, B. Pinter, J. Košmrlj, Nat. Commun. 2018, 9, 4814. doi: 10.1038/s41467-018-07081-5.
[4] J. He, M. Wasa, K. S. L. Chan, J. Yu, J. Am. Chem. Soc. 2013, 135, 3387. doi: 10.1021/ja400648w. [5] (a) C. Liu, C. Ji, X. Hong, M. Szostak, Angew. Chem. Int. Ed. 2018, 57, 16721. doi: 10.1002/anie.201810145.(b) C. Liu, C. Ji, T. Zhou, X. Hong, M. Szostak, Org. Lett. 2019, 21, 9256. doi: 10.1021/acs.orglett.9b03678.
(c) C. Liu, C. Ji, T. Zhou, X. Hong, M. Szostak, Angew. Chem. Int. Ed. 2021, 60, 10690. doi: 10.1002/anie.202100949.
(d) G. Meng, M. Szostak, Angew. Chem. Int. Ed. 2015, 54, 14518. doi: 10.1002/anie.201507776.
(e) C. Liu, M. Szostak, Angew. Chem. Int. Ed. 2017, 56, 12718. doi: 10.1002/anie.201707102.
[6] H. Lu, T. Yu, P. Xu, H. Wei, Chem. Rev. 2021, 121, 365. doi: 10.1021/acs.chemrev.0c00153.
















No comments yet.