
导读:
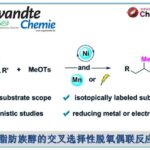
近日,厦门大学的霍浩华课题组在Nat. Chem.中发表论文,报道一种全新的金属光氧化还原催化的自由基方法,实现了N-烷基苯胺的α-C(sp3)–H芳基化反应。该方法引入了一种结构简单且具有位阻效应的芳基酮光催化剂,其关键创新在于有效抑制了不利的逆向电子转移过程,从而实现高效的α-anilinoalkyl自由基生成。该策略通过连续的单电子转移与质子转移过程,克服了现有方法的多种局限。结合手性镍催化剂,还可实现多种N-烷基苯胺与各类(杂)芳基卤的位点选择性、对映选择性芳基化反应。此外,该方法展现出卓越的官能团耐受性,能够对复杂分子的结构进行模块化官能团修饰。
Direct enantioselective C(sp3)−H coupling of N-alkyl anilines via metallaphotoredox catalysis
W. Zu, X. Wan, H. Wu, J. Huo, C. Zhang, C. Li, Y. Huang, Z. Xu, Y. Xu, T. Li, J. Cheng, J. Ye, C. Wang, H. Huo, Nat. Chem. 2026, ASAP. doi: 10.1038/s41557-025-02018-0.
正文:
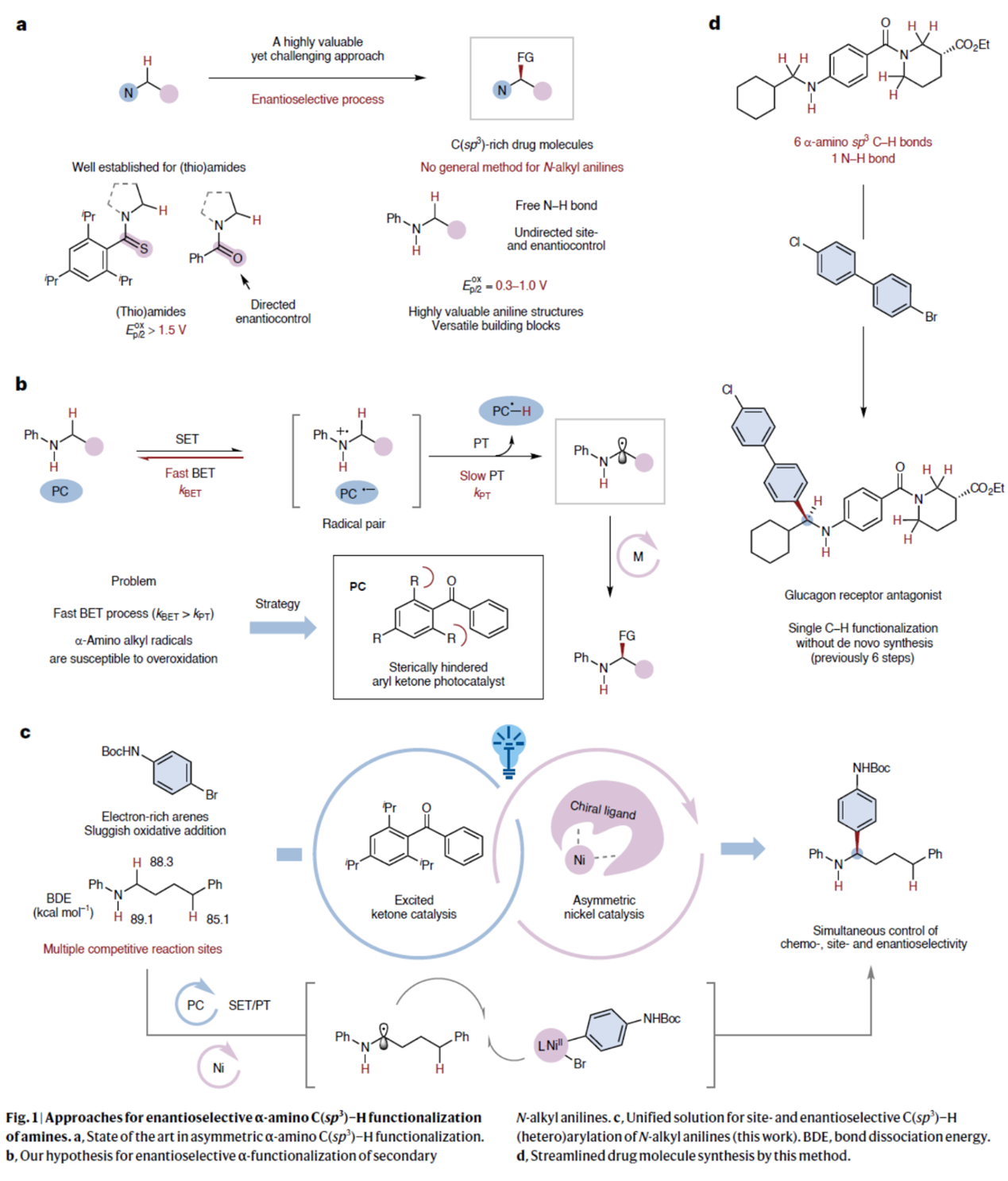
胺类化合物是药物分子、农用化学品及天然产物中普遍存在的结构单元。其中,N-烷基苯胺因其显著的生物活性以及作为后续可修饰多功能砌块的特性,成为一类重要结构单元。有机化学的核心目标在于开发直接胺基官能团化的高效策略,从而实现三维类药分子的合成。对映选择性α-氨基C(sp3)−H官能团化是实现具有增强三维结构特征、结构可调性及功能多样性的胺类化合物合成的理想途径[1](Fig. 1a)。然而,胺官能团(尤其含游离N-H键)的Lewis碱性对过渡金属催化过程构成显著挑战,因其易与催化剂配位并导致催化剂失活。因此,过渡金属催化胺类α-C(sp3)−H键官能团化仍具有挑战,尤其是在需要同时控制化学选择性与对映选择性的情况下。目前高效的不对称胺类α-C(sp3)−H键官能团化方法尚少[2],主要依赖N-保护(硫代)酰胺或特定环状叔胺结构。
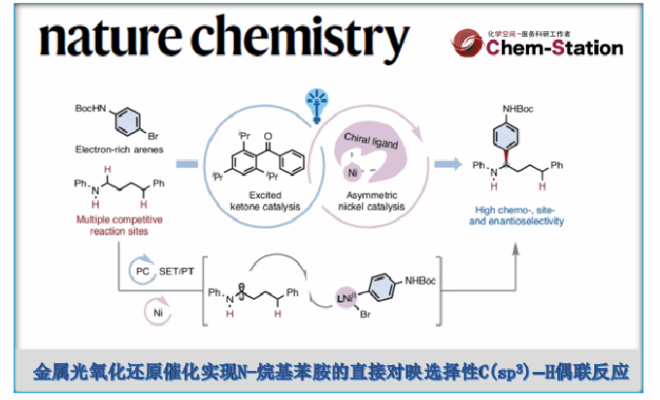
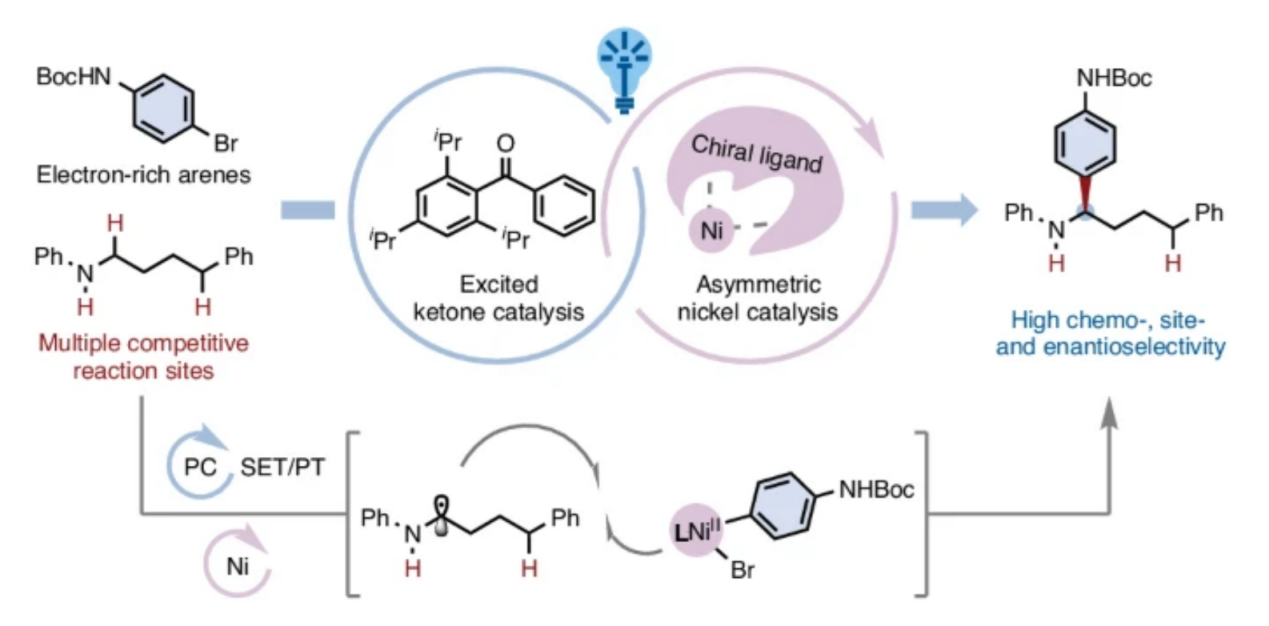
受这一基础性挑战的启发,霍浩华课题组探究了金属光氧化还原催化自由基策略能否实现二级N-烷基苯胺的α-C(sp3)−H键官能团化[3](Fig. 1b)。鉴于N-烷基苯胺通常具有较低氧化电势且本征适宜单电子氧化过程,霍浩华课题组致力于构建新型金属光氧化还原催化平台。该平台通过连续单电子转移(SET)-质子转移(PT)机制,实现了N-烷基苯胺的对映选择性α-C(sp3)−H键官能团化。鉴于氮原子与芳香环的共轭效应,该策略面临一个公认的挑战,即还原态光催化剂与二级苯胺自由基阳离子之间会发生快速逆向电子转移(BET)。为了克服此问题,霍浩华课题组提出假设:位阻型芳基酮光催化剂可延缓孪生自由基对间不利的BET过程。其增强的空间位阻效应、超共轭效应及给电子效应可稳定单电子转移(SET)后生成的羰基负离子自由基,从而延长其寿命并降低BET倾向,最终提升α-氨基烷基自由基的催化生成效率。
为展示这一概念独特的策略,霍浩华课题组选择采用镍金属光氧化还原催化体系,实现N-烷基苯胺与多种芳基卤的C(sp3)−H芳基化反应(Fig. 1c)。具体而言,霍浩华课题组设想三线态激发态酮类光催化剂可通过高选择性的单电子转移/质子转移(SET/PT)过程活化N-烷基苯胺,从而产生α-氨基烷基自由基。随后,该碳中心自由基与芳基溴在手性镍催化剂作用下发生对映选择性偶联,生成具有重要价值的胺类分子。因此,若能实现该策略,将开辟全新的逆合成分析路径,并无需从头合成即可加速从现有药物库中发现先导化合物(Fig. 1d)。
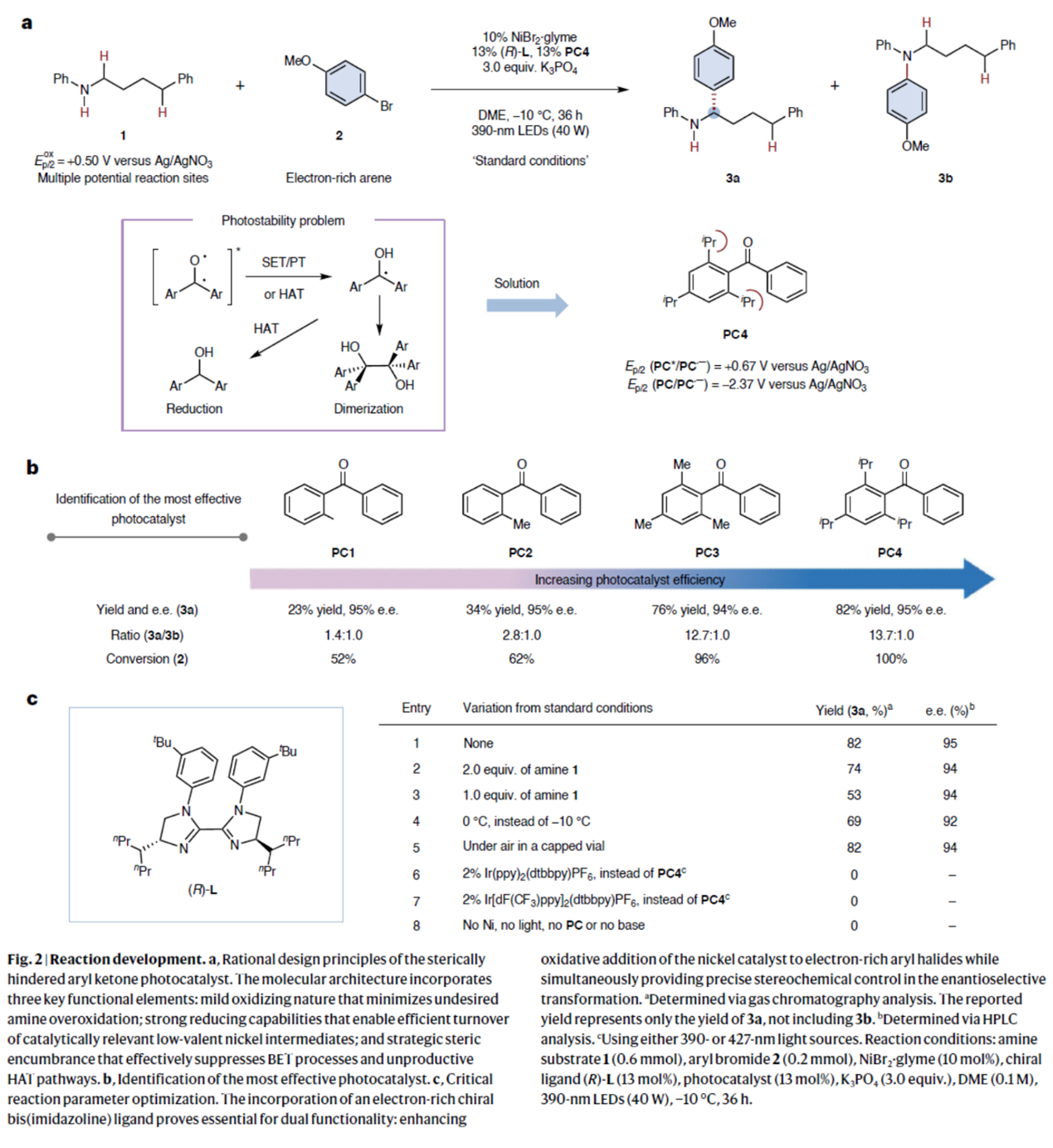
首先,作者采用N-烷基苯胺1与4-溴苯甲醚2作为模型底物,对反应的光催化剂进行了设计与筛选(Fig. 2)。进而确定最佳的反应条件为:采用NiBr2·glyme作为催化剂,(R)-L作为手性配体,PC4作为光催化剂,K3PO4作为碱,390 nm LEDs (40 W)作为光源,在DME反应溶剂中,反应温度为-10 oC,最终获得82%收率的产物3a(95% ee)。
在上述的最佳反应条件下,作者对一系列N-烷基苯胺的底物范围进行深入研究(Table 1)。研究结果表明,一系列非环状二级烷基胺或环状三级烷基胺,均可与N-Boc-4-溴苯胺顺利进行反应,获得相应的产物4–20,收率为58-93%,ee为84-98%。同时,一系列具有不同HAT敏感性C(sp3)−H键的N-烷基苯胺,也成功参与了反应,实现了优异的化学选择性、位点选择性和立体选择性,获得相应的产物21–39,收率为40-92%,ee为81-96% ee,dr高达99:1。
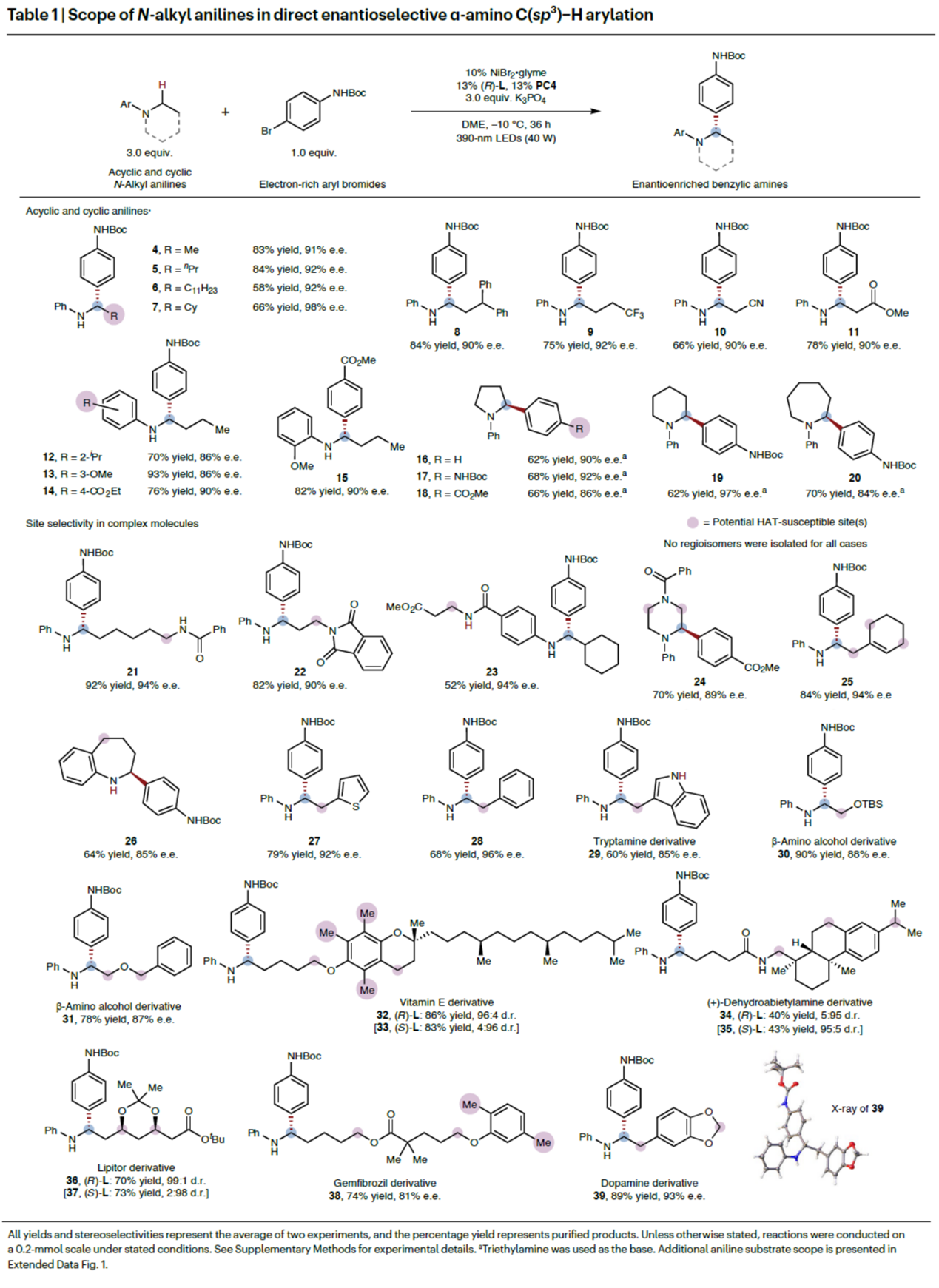
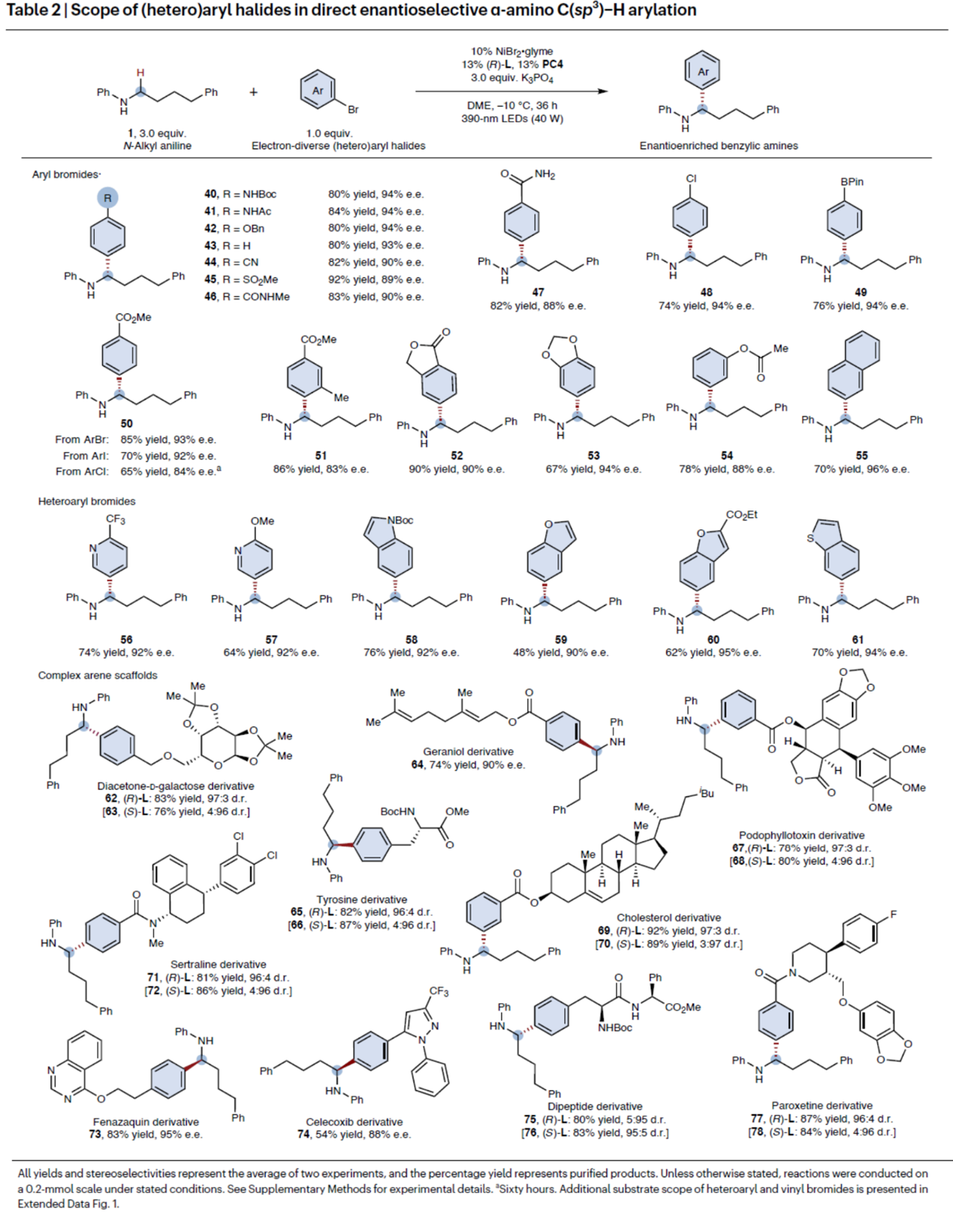
之后,作者还对(杂)芳基卤的底物范围进行了扩展(Table 2)。研究结果表明,一系列具有不同取代的(杂)芳基卤,均可与N-烷基苯胺1顺利进行反应,获得相应的产物40–61,收率为48-92%,ee为83-96%。其中,含有一系列活性基团的底物,如卤素、硼基、烷氧羰基等,均与体系兼容。值得注意的是,该策略还可用于药物、天然产物和生物分子的后期衍生化,获得相应的产物62–78,收率为54-92%,ee高达95%,dr高达97:3。上述结果表明,该催化方法能够利用简单原料和结构复杂的芳基卤化物制备出具有高对映选择性的复杂苄胺类化合物,同时凸显了该镍光氧化还原催化体系卓越的化学选择性、位点选择性和对映选择性。
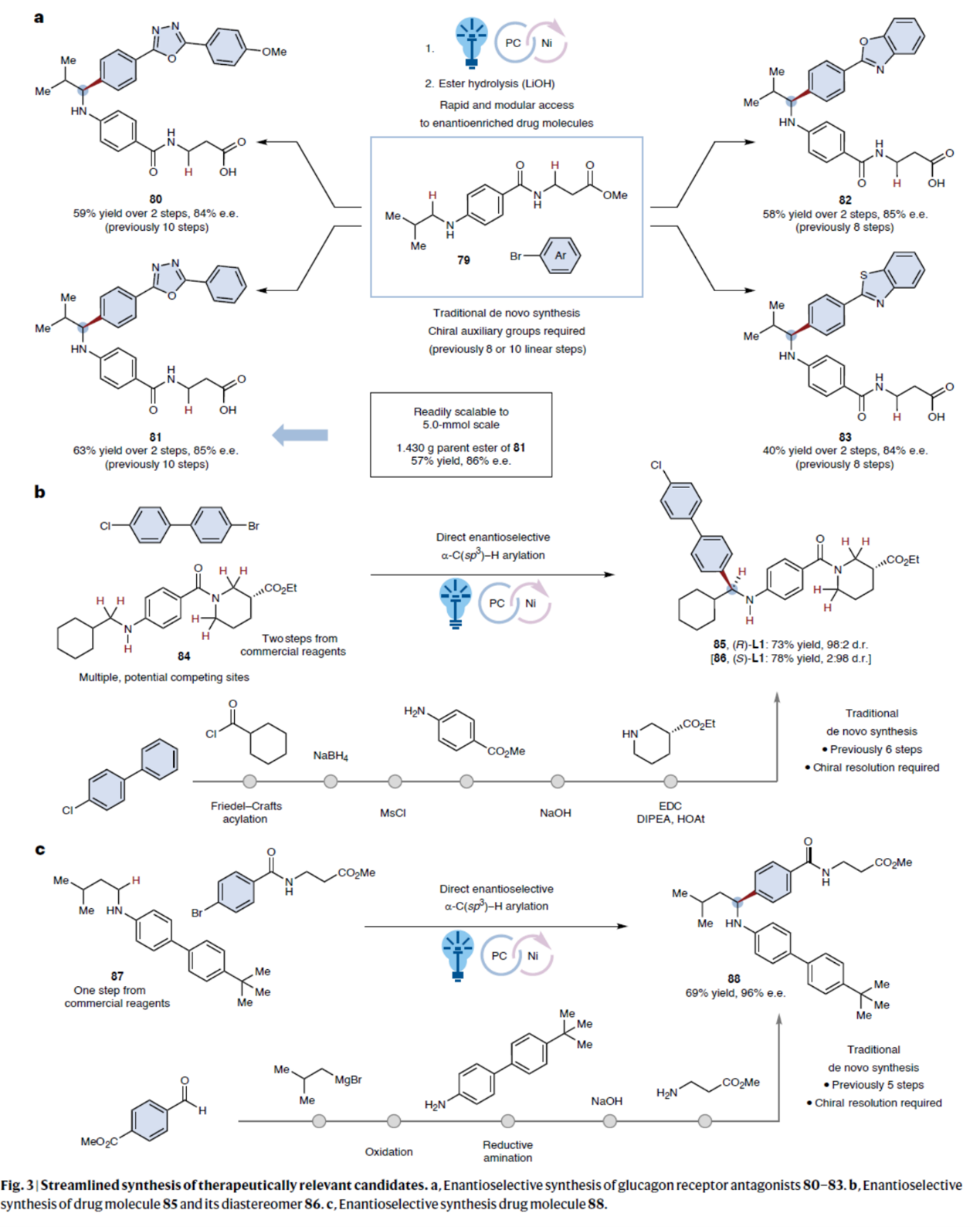
接下来,作者对反应的实用性进行了研究(Fig. 3)。首先,作为治疗糖尿病用胰高血糖素受体拮抗剂的手性苄胺化合物80–83,此前合成时需采用手性助剂策略,经历八至十步反应。相比之下,采用上述的新方法仅需两步即可模块化合成这四种分子(总收率40–63%,ee为84–85%)。该方法以单一原料N-烷基苯胺79与市售芳基溴为直接起始物料,通过高效的C–H芳基化/酯水解串联反应实现。其次,兼具胰高血糖素受体拮抗活性、可用于糖尿病预防或治疗的苄胺化合物85及其非对映异构体86,此前的合成需经历六步反应,且须对每个非对映异构体进行手性色谱拆分。采用上述的方法通过催化对映选择性合成技术,以N-烷基苯胺84与市售芳基卤为原料,一步实现两种非对映异构体的合成,并具有良好的收率与优异的立体选择性。此外,作为胰高血糖素受体拮抗剂的苄胺化合物88,其原工艺需五步合成,且须通过手性拆分才能获得两种对映异构体中的目标构型。采用上述的方法,可直接以简单N-烷基苯胺87为原料合成该分子,具有69%收率及96%ee。
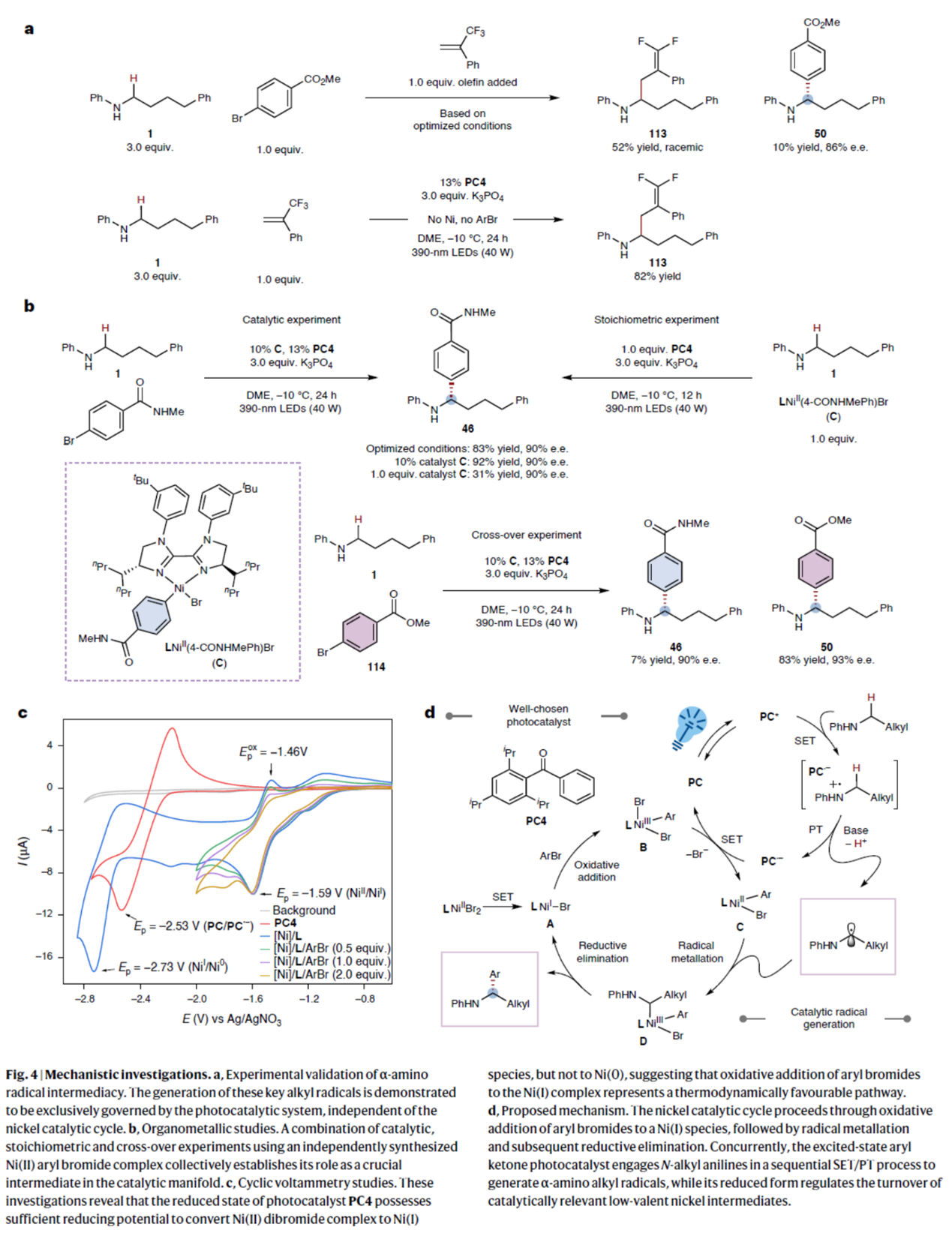
最后,作者对反应的机理进行了研究(Fig. 4)。首先,自由基捕获实验结果表明,反应形成了α-氨基烷基自由基。同时,这些关键烷基自由基的生成完全受光催化体系控制,与镍催化循环无关(Fig. 4a)。其次,通过对独立合成的芳基溴化镍(II)配合物C进行催化实验、化学计量实验及交叉实验,综合研究结果共同证明该配合物是催化循环中的关键中间体(Fig. 4b)。此外,CV实验结果表明,还原态光催化剂PC4具有足够的还原能力,可将二溴化镍配合物转化为Ni(I)物种,但不能还原至Ni(0)状态。因此,芳基溴化物与Ni(I)配合物发生的氧化加成反应是一条热力学上可行的途径(Fig. 4c)。基于上述的实验研究,作者提出如下合理的反应机理(Fig. 4d)。该过程始于二级苯胺与三重态光催化剂(PC*)之间的单电子转移(SET),形成离子-自由基对。在碱存在下,这些离子自由基物种通过质子转移(PT)生成α-氨基烷基自由基和羰基负离子自由基(PC·−)。同时,由镍(II)预催化剂通过羰基负离子自由基单电子还原产生的溴化镍(I)配合物A,与芳基溴发生氧化加成,形成二溴化芳基镍(III)配合物B。该配合物被羰基负离子自由基还原,再生成基态光催化剂和溴化芳基镍(II)配合物C。后者捕获氧化生成的烷基自由基,形成镍(III)配合物D。最终,经还原消除得到对映体富集的苄胺,并再生溴化镍(I)配合物A并进入后续催化循环。当前阶段,尽管不能完全排除潜在的镍(0)/镍(II)机理路径,但根据上述的机理研究,该可能性较低。
总结:
厦门大学的霍浩华课题组开发了一种稳健高效的N-烷基苯胺对映选择性α-氨基C(sp3)–H芳基化方法,该方法利用先进的金属光氧化还原催化体系。通过采用精心设计的位阻芳基酮光催化剂与富电子手性双咪唑啉配体,该策略解决了胺官能团化领域长期存在的难题。本方法展现出宽泛的底物适用性,可兼容多种N-烷基苯胺与芳基溴,并实现优异的化学选择性、位点选择性和立体选择性。
参考文献:
[1] F. Lovering, J. Bikker, C. Humblet, J. Med. Chem. 2009, 52, 6752. doi:10.1021/jm901241e.
[2] P. Jain, P. Verma, G. Xia, J. Q. Yu, Nat. Chem. 2017, 9, 140. doi:10.1038/nchem.2619.
[3] N. Holmberg-Douglas, D. A. Nicewicz, Chem. Rev. 2022, 122, 1925. doi:10.1021/acs.chemrev.1c00311.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
关注Chem-Station抖音号:79473891841
请登陆TCI试剂官网查看更多内容
https://www.tcichemicals.com/CN/zh/



















No comments yet.