
作者:陈才友课题组
导读:
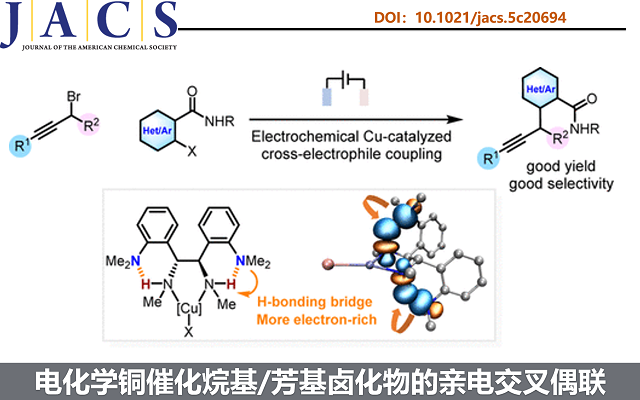
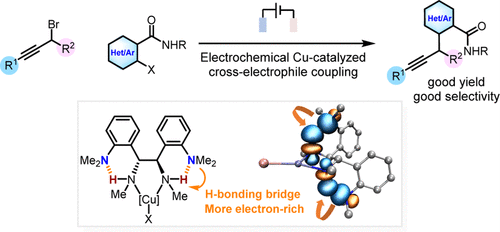
近日,武汉大学陈才友教授课题组《Journal of the American Chemical Society》上,发表了题为“Expanding Ullmann Homocoupling to Cross-Coupling: Electrochemical Copper-Catalyzed Cross-Electrophile Coupling of Alkyl and Aryl Halides”的论文。该团队设计了一种配体促进的电化学铜催化亲电交叉偶联策略,成功实现了首例电化学铜催化的烷基与芳基卤化物交叉亲电偶联反应。陈才友教授为论文通讯作者,博后梁凯伦为该论文的第一作者。
“Expanding Ullmann Homocoupling to Cross-Coupling: Electrochemical Copper-Catalyzed Cross-Electrophile Coupling of Alkyl and Aryl Halides
Kailun Liang, Yuhongxu Bai, Hang Li, Yingjie Li, Chengbiao Zhu, Zhenwei Wei, Caiyou Chen*
J. Am. Chem. Soc.2026, 148, 7, 7472–7481. DOI :10.1021/jacs.5c20694
正文:
碳-碳键的构建是有机合成化学中的中心转化。传统的Ullmann型反应和Hurtley反应虽然能够实现C(sp³)-C(sp²)键的构建,但分别依赖于有机金属试剂或具有活性亚甲基的底物,这些严格要求大大限制了其实际应用范围(图1a)。尽管铜作为首个用于C-C键构建的金属催化剂具有成本低廉、毒性低以及易于促进挑战性化学键形成等优势,但现代交叉亲电偶联反应领域长期被镍催化所主导,铜催化的烷基卤化物与芳基卤化物交叉亲电偶联反应始终未能实现。这一空白主要源于两大挑战:化学选择性控制(抑制自偶联,实现交叉偶联)以及Cu(I)与有机卤化物缓慢的氧化加成过程(图1b)。有机电化学作为一种可扩展且可持续的合成平台,能够精确调控过渡金属配合物的氧化还原状态而无需化学计量的氧化剂或还原剂,为解决上述难题提供了新的契机。
近日,陈才友教授团队设计了一种配体促进的电化学铜催化亲电交叉偶联策略。该策略的核心设计包含两个关键要素:首先,选用炔丙基卤化物作为偶联伙伴之一,利用其与Cu(I)发生卤素原子转移(XAT)的速率高于芳基卤化物的特性,通过生成相对稳定的炔丙基/联烯基铜中间体来维持较低的自由基浓度,从而抑制均偶联副反应(图1c);其次,针对氧化加成缓慢的问题,团队创新性地设计了邻二甲氨基取代的二胺配体(L1),利用分子内N-H···N氢键作为桥梁促进NMe₂基团与配位氮原子之间的超共轭效应,显著增强配体的给电子能力,从而促进Cu(I)与亲电试剂的顺利反应,同时通过降低Cu(I)物种的还原电位来抑制阴极铜沉积导致的催化剂失活(图1d)。
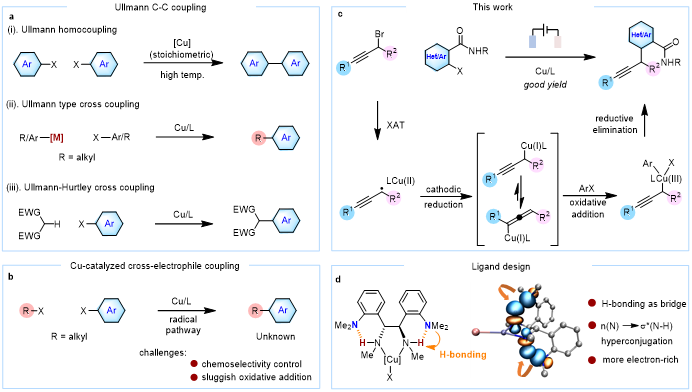
图1. 研究背景及本文策略
该研究团队以炔丙基溴1和2-碘苯甲酰胺2为模型底物,系统考察了反应条件。控制实验表明,无电流、无配体L1或无铜催化剂时反应几乎不发生;反应对氧气不敏感(空气中产率70%),但对水较为敏感;使用Mg、Zn或Mn粉末代替电化学还原则无法获得产物,表明电化学还原在还原高价铜物种方面具有独特优势。配体筛选揭示了显著的结构-活性关系:邻位二甲氨基配体L1表现最优(产物3/4/5的产率分别为74%/29%/10%),而去除NMe₂基团(L2)或将其移至对位(L3)均导致产率大幅下降;甲氧基取代的L4虽能获得中等产率,但移至对位(L5)后显著降低;引入吸电子CF₃基团(L6)则几乎使反应失效。双膦配体(L14-L15)和BINOL类配体(L16)完全失活,凸显了该二胺配体体系的独特优势(图2)。
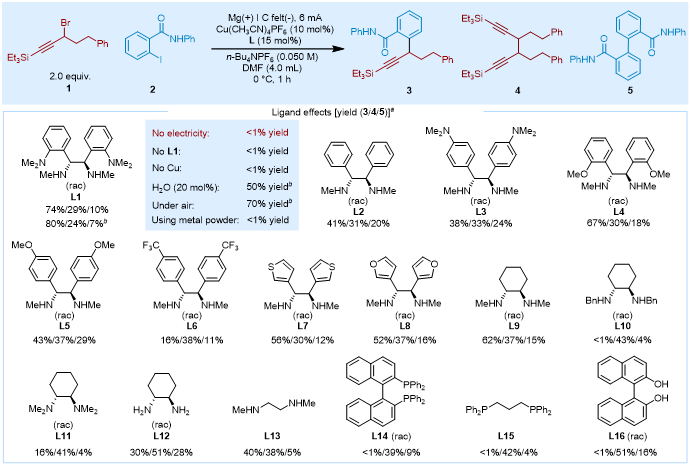
图2. 条件筛选
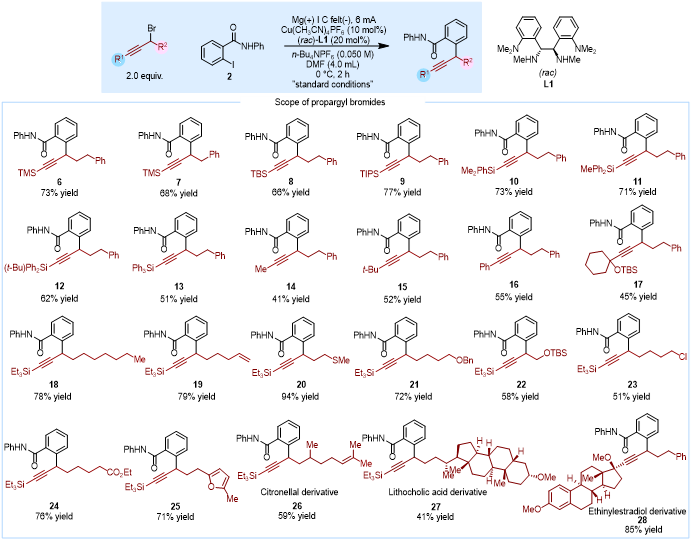
图3. 炔丙基卤代物拓展

图4. 芳基卤代物拓展
在最优条件下,研究展示了优异的底物普适性。对于炔丙基溴组分,各种硅基取代基(TMS、TBS、TIPS、Et₃Si、Me₂PhSi、MePh₂Si、(t-Bu)Ph₂Si、Ph₃Si)均能良好兼容(产率51-77%),烷基和苯基取代的炔烃也能中等产率获得产物;烷基链上可容纳烯烃、醚、硫醚、卤素、酯基、呋喃等多种官能团;更有价值的是,该反应适用于香茅醛、石胆酸、炔雌醇等天然产物和药物衍生的炔丙基溴(产率41-85%,图3)。对于芳基卤化物,除碘代物外溴代物同样适用;苯甲酰胺导向基团的N-芳基取代基可耐受多种电子效应和位阻效应的取代基,甚至强配位的8-氨基喹啉和皮考啉酰胺基团也几乎不影响产率;未保护的2-碘苯甲酰胺、烷基酰胺取代基均能顺利反应;芳环上的给电子基(甲氧基、甲基)和吸电子基(氟、氯、三氟甲基)均兼容,萘环、吡啶、噻吩等杂环也能参与反应。此外,氨基酸、萘普生、L-薄荷醇、胆固醇等复杂生物活性分子衍生的底物均能获得良好产率(39-88%),展示了该方法在复杂分子合成中的实用价值(图4)。
该研究不仅实现了方法学的突破,更重要的是为复杂药物分子的后期修饰提供了新工具。通过将生物活性分子(如萘普生、L-薄荷醇、胆固醇)转化为芳基卤化物底物,研究团队成功实现了这些分子的C(sp³)-C(sp²)键构建,为药物化学中的结构多样性合成开辟了新途径。特别是该方法对未保护官能团(如游离酰胺、氨基)的良好兼容性,大大简化了合成路线设计,减少了保护基操作步骤,符合现代绿色化学和步骤经济性的要求。此外,反应条件温和(0°C)、无需严格无水无氧操作,为其在工业规模应用提供了可能。
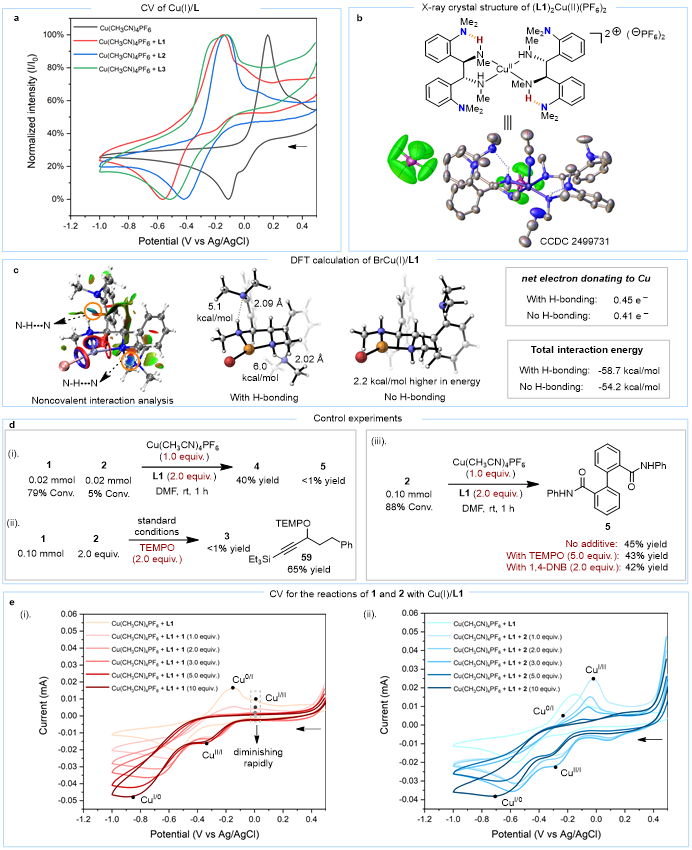
图5. 机理研究
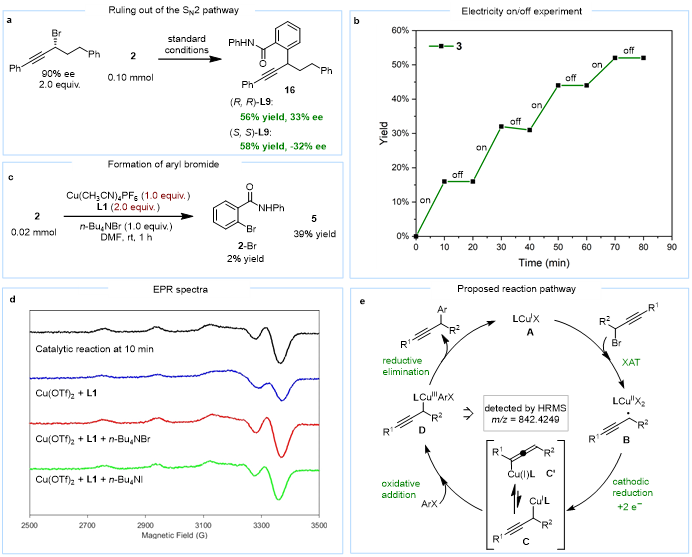
图6. 机理研究
作者通过机理研究系统验证设计假设。循环伏安法(CV)研究表明,L1/Cu(I)相比L2和L3具有更负的还原电位(分别低0.14V和0.05V),证实其更不易发生金属沉积(图5a)。X射线晶体学和DFT计算证实了L1Cu(II)配合物中存在N-H···N氢键,该氢键使Cu(I)配合物能量降低2.2 kcal/mol,氢键键能约5-6 kcal/mol(图5b)。电荷分解分析显示,有氢键时配体向金属中心的电子转移从0.41e⁻增至0.45e⁻,相互作用能从-54.2 kcal/mol增强至-58.7 kcal/mol(图5c)。控制实验表明炔丙基溴1与Cu(I)的反应速率(79%转化率)远快于芳基碘2(5%转化率);TEMPO捕获实验获得65%产率的炔丙基自由基加合物,而芳基碘与Cu(I)的反应不受TEMPO或电子捕获剂1,4-DNB影响,表明前者经自由基路径而后者经协同氧化加成路径(图5d)。手性转移实验排除了SN2机理(图6a)。EPR检测到Cu(II)物种,原位阶梯扫描伏安流式电化学高分辨质谱检测到Cu(III)中间体(m/z = 842.4249)(图6d)。DFT计算支持XAT步骤能垒(10.6 kcal/mol)低于协同氧化加成(12.2 kcal/mol),阴极还原生成炔丙基铜(I)后,与芳基碘的氧化加成能垒为7.8 kcal/mol,最终还原消除能垒为7.8 kcal/mol,整个催化循环热力学上高度有利(图7)。
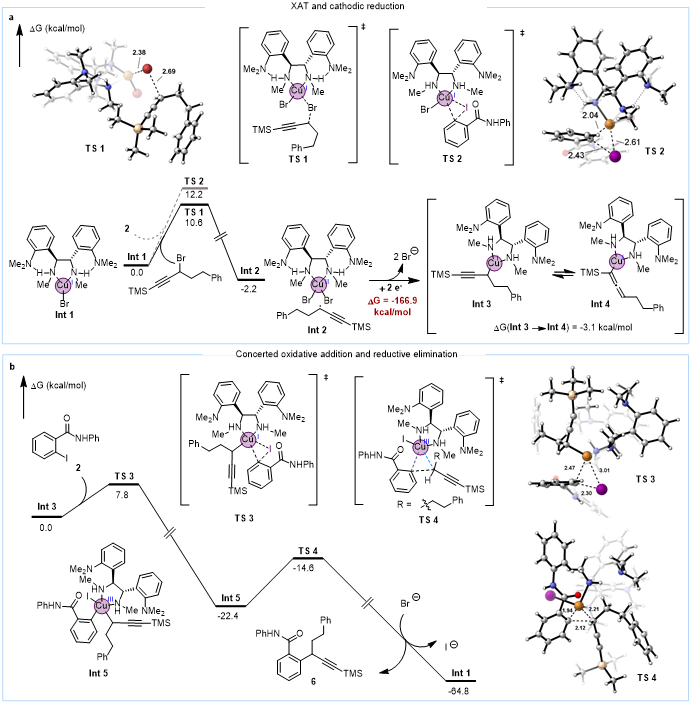
图7. DFT计算
该研究工作得到了国家重点研发计划(2023YFA1508600)、国家自然科学基金(22571239)、中央高校基本科研业务费(2042024kf1016)、武汉市自然科学基金探索(晨光)计划(2024040801020226)及武汉大学启动经费的支持。
陈才友教授简介
陈才友,武汉大学化学与分子科学学院教授,博士生导师,课题组长,国家重点研发计划青年项目首席科学家。2022年入选国家海外高层次人才引进计划,同年入选湖北省海外高层次人才项目,2023年入选武汉英才。2012年本科毕业于武汉大学,2017年博士毕业于武汉大学,师从张绪穆教授。2017~2022年,在美国加州理工学院从事博士后研究,师从美国科学院院士Gregory C. Fu和Jonas C. Peters。迄今在Nature (2篇); J. Am. Chem. Soc.; Angew. Chem. Int. Ed.; Acc. Chem. Res.; Chem. Sci.等杂志上发表论文30余篇,同时授权5项专利。2017年,受邀参加第67届德国Lindau诺贝尔奖获得者大会;2022年,受邀参加第四届“世界顶尖科学家大会”。曾荣获2017年度“武汉大学十大学术之星”称号。2025年荣获“新和成”-《中国化学》创新奖。


















No comments yet.