
作者:石油醚
引言
癫痫是一种由中枢神经系统异常引起的慢性脑部疾病,表现为无诱因的癫痫发作、行为或感觉异常,有时伴随意识丧失。全球约有5000万人患病,是世界上最常见的神经系统疾病之一,其中印度约1000万。据世卫组织(WHO)数据,规范诊断与治疗可使近70%患者实现无发作。然而,在低收入国家,因诊疗可及性差、疾病污名化、临床经验不足及卫生系统优先级低,癫痫相关死亡持续上升。但是,使用经济有效的抗惊厥药物是可行且关键的干预手段。拉考沙胺(R-Lacosamide)是一种新型N-甲基-D-天门冬氨酸(NMDA)受体甘氨酸位点结合拮抗剂,被誉为第三代”抗癫痫药物。剂型包括薄膜衣片、糖浆、注射液及口服溶液,其中口服溶液适用于4岁及以上患者的联合治疗。
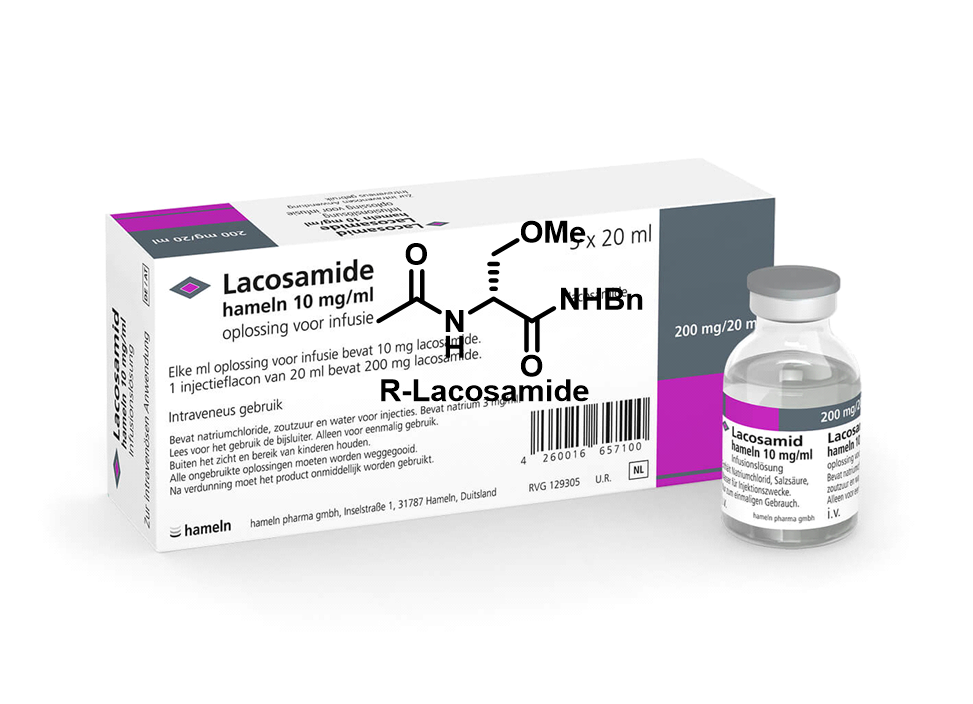
图1拉考沙胺(Lacosamide,LCM)
拉考沙胺(Lacosamide,LCM):前世今生
拉考沙胺(R-Lacosamide)是由比利时优时比公司(UCB Pharma)的德国子公司SchwarzBioSciences公司研发的一种新型N-甲基-D-天门冬氨酸(NMDA)受体甘氨酸位点结合拮抗剂。其可以选择性增强钠通道缓慢失活,减少神经元兴奋性。该药于2008年9月、2009年5月和2020年相继在欧盟、美国和中国上市,用于癫痫部分性发作的单药或联合治疗,以及脑肿瘤患者的辅助治疗。(R-Lacosamide)—— (R)-2-acetamido-N-benzyl-3-methoxypropionamide——已成为合成化学研究热点,已有多种不对称合成、手性池衍生及酶催化路线被报道。合成1的方法可根据手性引入方式分为三类:手性池法、不对称合成法和拆分法。
拉考沙胺(Lacosamide,LCM)的合成路线
1、[手性池法]
1.1 D-Serine (2)作为原料
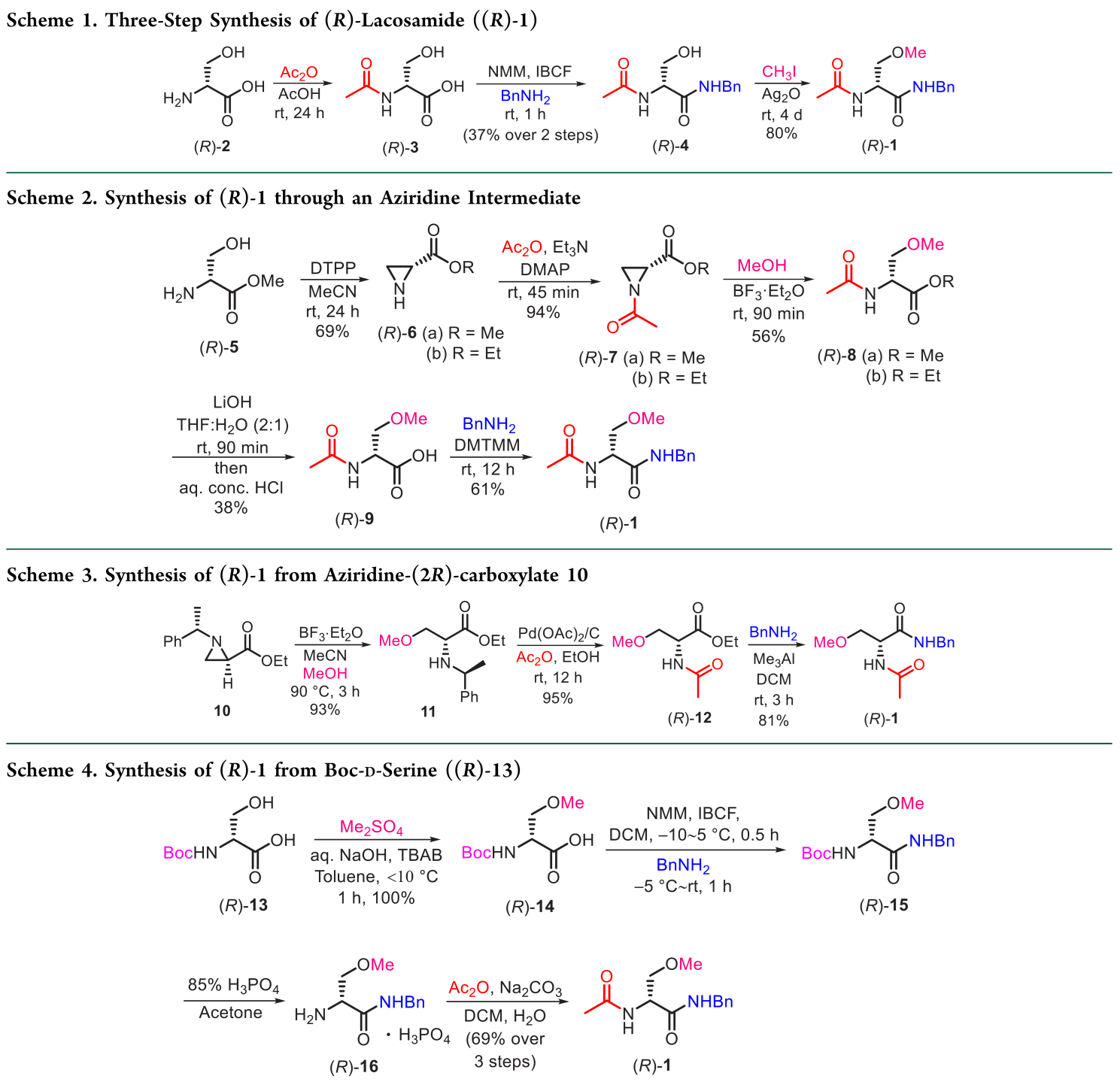
早期药物合成以非天然氨基酸D-丝氨酸((R)-2)为起始原料,经N-酰化和O-甲基化构建(R)-1;保护基策略因步骤顺序不同而各异。其中O-甲基化易导致部分消旋。为抑制消旋,Choi等人采用MeI/Ag₂O中性甲基化法,三步完成(R)-2→(R)-1转化(30% yield)。但该法需过量Ag₂O(5 equiv),试剂昂贵、不可回收,且甲基化耗时长达4天(Scheme 1)。
1.2 D-Serine Methyl Ester (5)作为原料
以D-丝氨酸甲酯((R)-5)为起始物:经DTPP环化得氮丙啶(R)-6;酰化后,以BF₃·Et₂O催化甲醇开环得(R)-8;再水解得酸(R)-9;最后在DMTMM介导下与苄胺偶联,五步合成(R)-1 (8.4% yield)(Scheme 2)。
1.3 Aziridine-(2R)-carboxylate (10) 作为原料
以市售Aziridine-(2R)-carboxylate (10)为起始物,经BF₃·Et₂O催化甲醇区域选择性开怀(94:6 dr);Pd(OH)₂/乙酸酐的作用下(R)-11发生乙酰化脱苄;Me₃Al 催化(R)-12 与苄胺偶联和经重结晶,三步以71%总产率和>99.9% ee获得(R)-1(Scheme 3)。
1.4 Boc-D-serine (13) 作为原料
Yang以以市售 Boc-D-丝氨酸((R)-13)作为起始物料(Scheme 4),经过四步以69%的产率、99.91% ee和100% 化学纯度合成(R)-1。具体路线是:(1)(R)-13 在 NaOH/TBAB 催化下与 (CH₃)₂SO₄ 发生甲基化,高选择性生成 (R)-14(>95% ee, >90% chemical purity),避免 Ag₂O 的使用并显著抑制消旋;(2)(R)-14 经异丁基氯甲酸酯/N-甲基吗啉活化后,与苄胺缩合得 (R)-15( 95.33% ee, 94.72%chemical purity);(3)(R)-15 用 85% H₃PO₄ 脱除 Boc ,直接沉淀为磷酸盐 (R)-16,ee 提升至 98.76%,化学纯度达 100%,且 12 个月内 ee 无下降;(4)(R)-16 经乙酸酐乙酰化即得目标物 (R)-1。该路线的核心优势在于起始原料 (R)-13 已含目标分子所需构型的 C2 手性中心;唯一局限是甲基化步骤存在微量不可控消旋及少量副产物。
1.5 L-serine (S)-2 作为原料
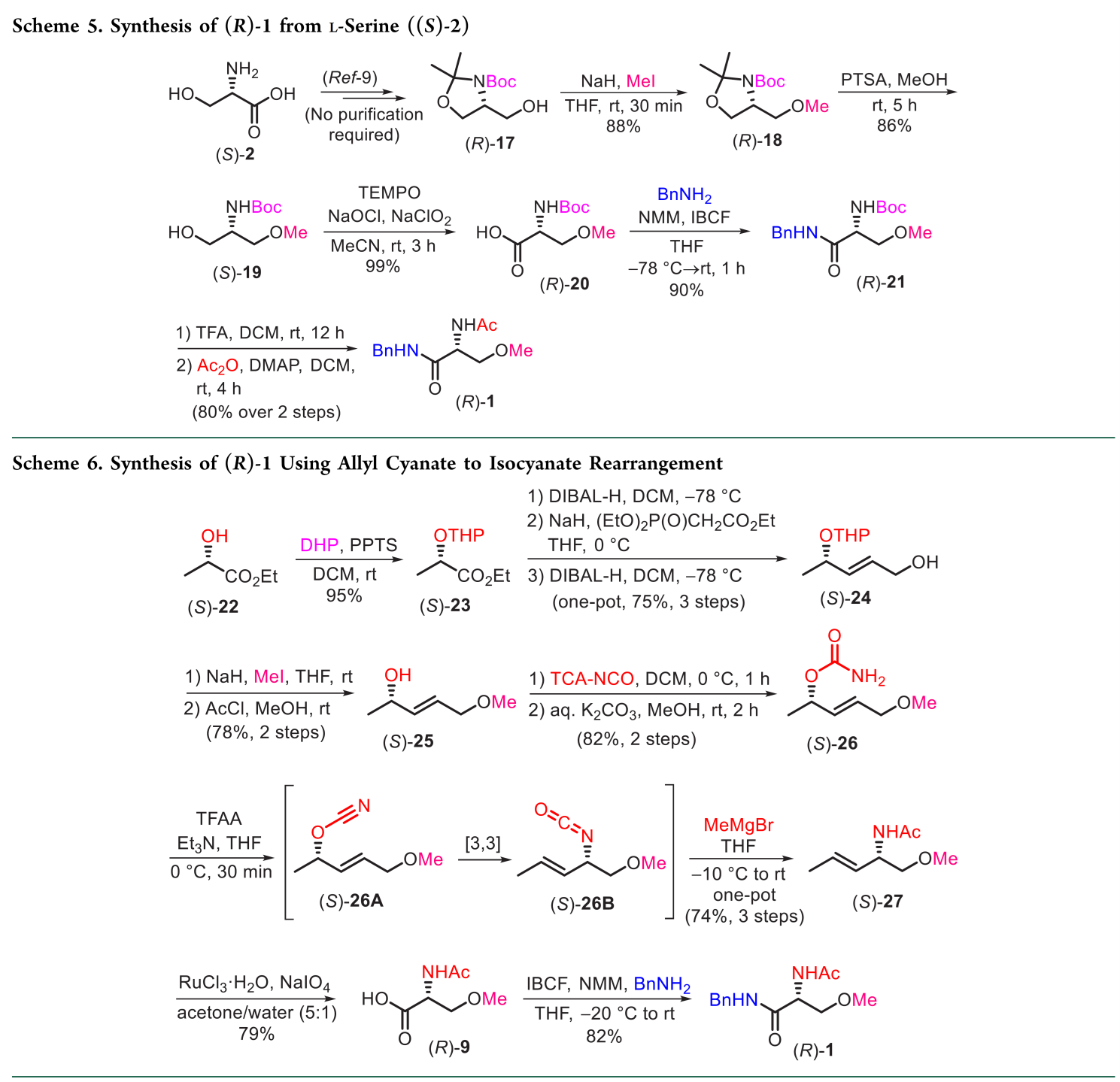
以天然氨基酸L-丝氨酸((S)-2)作为原料以44%的总产率获得(R)-1。即(S)-2经四步、无需中间体纯化,以81%总收率和克级规模制得N-Boc-N,O-异丙叉-L-丝氨醇 (R)-17);(R)-17在NaH/MeI条件下甲基化得(R)-18,全程无消旋(ee保持100%),故工业上更易得的(CH₃)₂SO₄或(CH₃O)₂CO亦可替代;(R)-18经酸性脱缩酮/TEMPO氧化,直接得到酸(R)-20(粗品用于下一步);(R)-20与苄胺经NMM/异丁基氯甲酸酯混合酸酐法偶联得酰胺(R)-21;最后,TFA脱Boc后乙酰化,即得(R)-1。全程手性中心稳定,终产物100% ee(Scheme 5)。
1.6 Ethyl L-Lactate (22) 作为原料
以L-乳酸乙酯((S)-22)为起始物,经七步合成(R)-1(22% yield,Scheme 6),核心步骤为立体专一性烯丙基氰酸酯→异氰酸酯重排。具体路线如下:(1)OTHPO保护的(S)-23经DIBAL-H还原得醛,不经纯化直接进行HWE烯化,生成α,β-不饱和酯;(2)该酯再经DIBAL-H还原、甲基化及THP脱保护,得烯丙醇(S)-25;(3)(S)-25经TCA-NCO酰化/水解得烯丙基氨基甲酸酯(S)-26,再经TFAA脱水生成烯丙基氰酸酯(S)-26A,其自发[3,3]重排为异氰酸酯(S)-26B;(4)(S)-26B与过量MeMgBr反应,高收率得N-乙酰化烯丙胺(S)-27;(5)(S)-27经RuCl₃/NaIO₄氧化裂解双键得酸(R)-9;(6)(R)-9经NMM/异丁基氯甲酸酯活化后与苄胺偶联得(R)-1。全程未发生消旋。该路线主要缺点是步骤长(七步)、收率低(22%),且依赖多种高危(DIBAL-H、MeMgBr)及昂贵试剂(RuCl₃·H₂O、TCA-NCO、TFAA)。
2、[不对称合成法]
2.1 Jacobsen’s Hydrolytic Kinetic Resolution
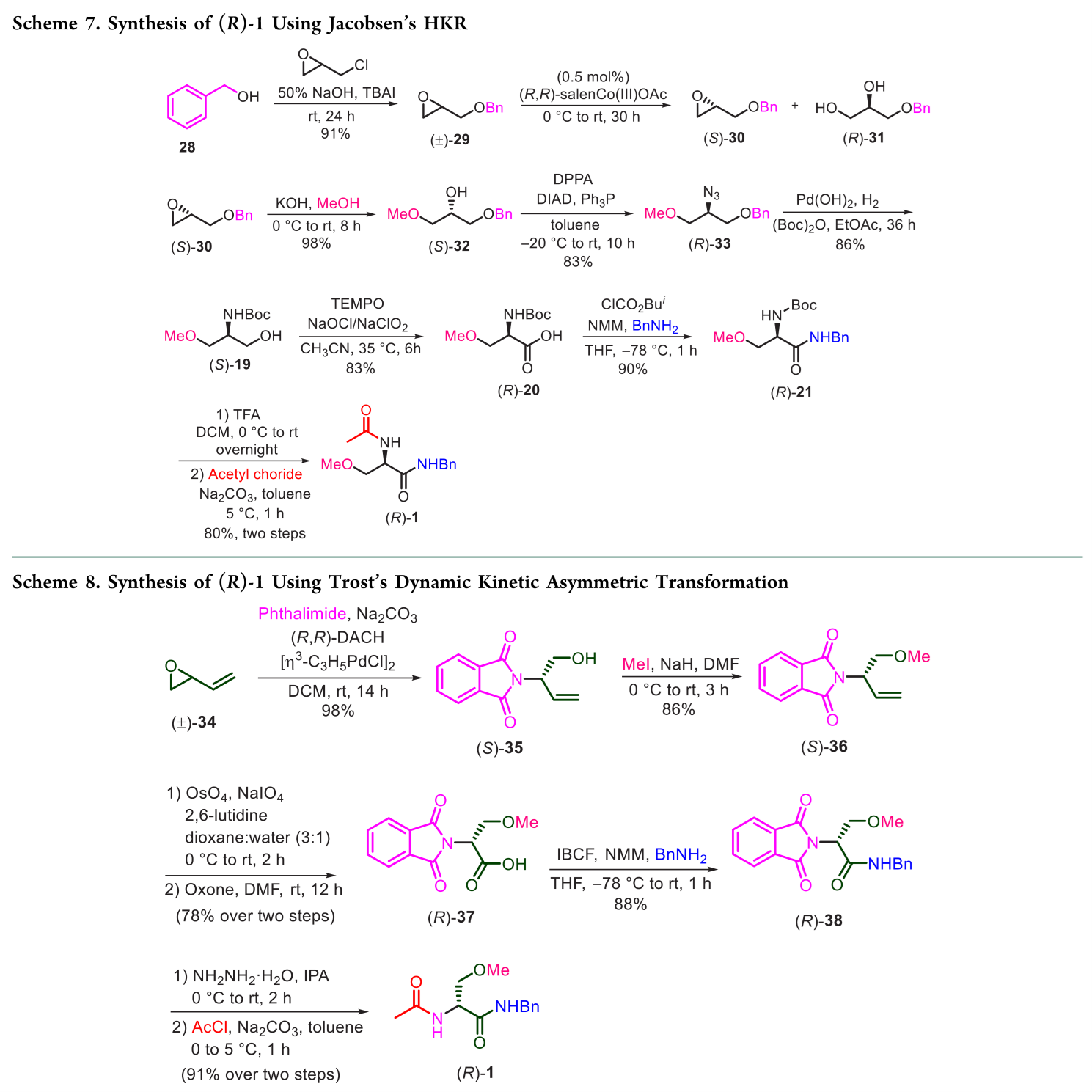
Muthukrishnan等人利用Jacobsen’s水解动力学拆分(HKR)以消旋苄基缩水甘油醚((±)-29)为底物,经(R,R)-salen-Co(III)OAc(0.5 mol%)催化、水解,高选择性获得(S)-30(47%收率,ee >99%)(Scheme 7)。随后:(1)(S)-30经甲醇区域选择性开环得仲醇(S)-32;(2)(S)-32与DPPA经Mitsunobu反应转化为叠氮化物(R)-33;(3)(R)-33在Pd(OH)₂/(Boc)₂O条件下氢化得N-Boc保护伯醇(S)-19;(4)(S)-19经TEMPO氧化为酸,再与苄胺偶联得酰胺(R)-21;(5)(R)-21经TFA脱Boc后乙酰化,即得(R)-1(18% overall yield)。该路线主要缺点是HKR步骤理论最大收率为50%(导致47%收率已接近极限),且依赖昂贵的Jacobsen手性催化剂。
2.2 Dynamic Kinetic Asymmetric Transformation.
Garg 和Pandey等利用Trost’s 动态动力学不对称转化(DYKAT)为关键步骤,以rac-butadiene monoepoxide(±)-34为原料,经(R,R)-DACH配体催化,一步高选择性制得对映纯邻苯二甲酰醇衍生物(S)-35;随后经四步官能团转化,五步总收率52%,即得(R)-1(Scheme 8)。该路线主要局限在于依赖昂贵的(R,R)-DACH配体,导致生产成本过高,难以实现(R)-1的商业化生产。
2.3 Sharpless Asymmetric Dihydroxylation
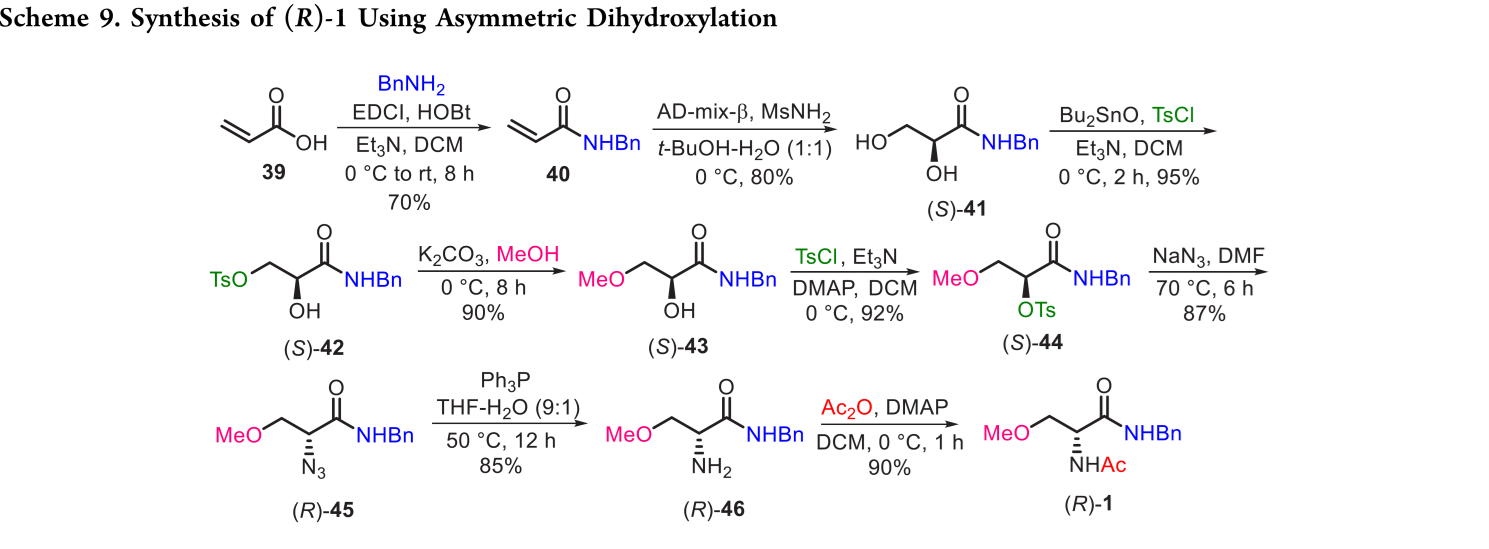
Narsaiah等人以丙烯酸(39)为原料,经苄胺保护得N-苄基丙烯酰胺(40),再经AD-mix-β催化的Sharpless Asymmetric Dihydroxylation(关键步骤)高选择性制得二醇(S)-41;后续经五步官能团转化,全程八步,总收率29%,即得(R)-1(Scheme 9)。
2.4 DYKAT策略
Imahori 等人利用动态动力学不对称转化(DYKAT)合成 1:环氧化物 38 与琥珀酰亚胺 39 在 π-烯丙基氯化钯二聚体/(R,R)-DACH(Trost 萘基配体)催化下反应,高选择性生成(S)-40;其羟基在 CF₃SO₃H 催化下与苯甲基三氯乙酰亚胺反应得(S)-41(NaH/BrCH₂Ph 法产率较低);(S)-41 经两步还原得酰胺(S)-42;后者在 H₂/Pd(OH)₂ 下氢化–氢解得醇(S)-8;再经 NaOCl–NaClO₂/TEMPO(cat.)氧化为酸(S)-9;最后与异丁基氯甲酸酯及 NH₃·H₂O 缩合,以 34% 总收率、>99% ee 获得 1(Scheme 16)。因需使用昂贵的(R,R)-DACH 配体,该路线不具工业可行性。
2.5 Ugi Multicomponent Reaction.
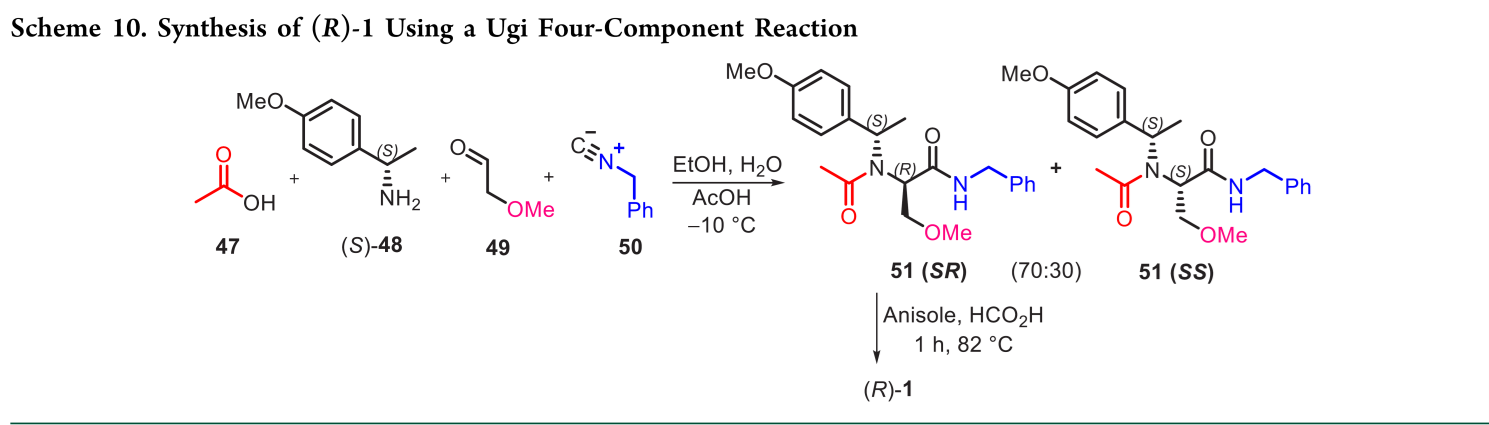
Wehlan 利用Ugi反应以(S)-1-(4-甲氧基苯基)乙胺((S)-48)为手性辅基合成(R)-1。苄基异氰酸酯(50)由苄胺现场制备;四组分(47–50)在−10 °C经Ugi反应,一步高收率生成非对映体混合物51(SR/SS = 70:30),其中目标产物51(SR)可直接分离;脱除手性辅基后,从苄胺出发,五步总收率40%,即得(R)-1,化学纯度与光学纯度均 >99.9%(Scheme 10)。该路线主要缺点在于:(1)依赖昂贵的手性辅基(S)-48;(2)Ugi反应产生30%非目标非对映体,降低原子经济性并增加分离负担;(3)脱辅基后存在手性杂质残留风险。
2.6 Asymmetric Hydrogenation Using a Chiral Rhodium Complex
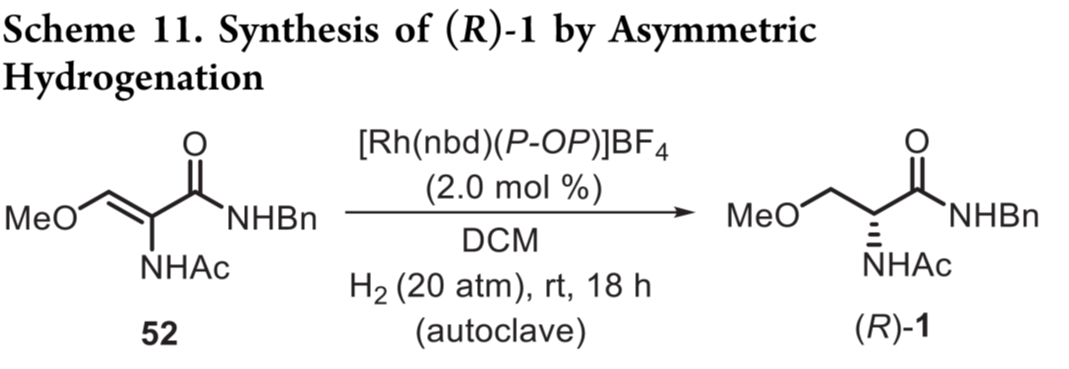
Etayo等人利用手性铑催化剂[Rh(nbd)(P-OP)]BF₄,在二氯甲烷中对β-烷氧基取代的α-(酰氨基)丙烯酰胺(52)进行不对称氢化,一步高收率、高选择性地直接得到(R)-1(99% ee),无其他异构体生成(Scheme 11)。
3、[拆分法]
3.1 Resolution of a Diastereomeric Mixture.
Bologna等人采用手性拆分法合成(R)-1(Scheme 12):乙酰基DL-丝氨酸甲酯((±)-53)与苄胺缩合得(±)-4(84%);(±)-4经NaOH/对甲苯磺酸甲酯甲基化得(±)-1(73%);(±)-1经稀HCl脱乙酰,再与(S)-2-氯扁桃酸成盐,两步总收率84%,生成非对映体混合物54(RS/SS);该混合物于EtOAc/EtOH中热溶后缓慢冷却至20 °C结晶,选择性析出54(RS)(37%);54(RS)经乙酸酐处理即得(R)-1(88%)。全程六步,总收率16.7%,起始原料为(±)-53。
3.2 Chemoenzymatic Method.
wang等人利用酶拆分法高效合成(R)-1(>98% ee,Scheme 13):DL-3-甲氧基丙氨酸((±)-55)经乙酸酐乙酰化得(±)-9;固定化大肠杆菌氨基酰酶(EC 3.5.1.14)选择性水解其L-乙酰基,一步分离获得高纯(R)-9(>99% ee)和(S)-55(>97% ee);(R)-9再与苄胺缩合,即得(R)-1(>98% ee)。
总结:
本综述系统综述了(R)-拉科酰胺的合成路线及其优劣。基于D-丝氨酸或其衍生物(如Boc-D-丝氨酸)的方法优势在于底物自带目标手性中心,但甲基化过程存在部分消旋及微量杂质;L-丝氨酸或L-乳酸乙酯路线虽可高选择性构建手性中心,却步骤冗长。Jacobsen HKR、Trost DYKAT、Sharpless二羟基化及手性Rh催化氢化等不对称合成法受限于贵金属催化剂成本,难以放大。消旋体拆分与化学酶法拆分虽可行,但理论最大收率仅50%,显著拖累整体效率。综合来看,丝氨酸类路线在原料成本、步骤经济性与工艺稳健性上优势突出,最具商业化潜力。
参考资料
A Short Review of Synthetic Routes for the Antiepileptic Drug (R)‑Lacosamide
Eswar K. Aratikatla†,‡ and Asish K. Bhattacharya
Org. Process Res. Dev. 2020, 24, 17−24. DOI: 10.1021/acs.oprd.9b00373




















No comments yet.