
本文作者:杉杉
导读
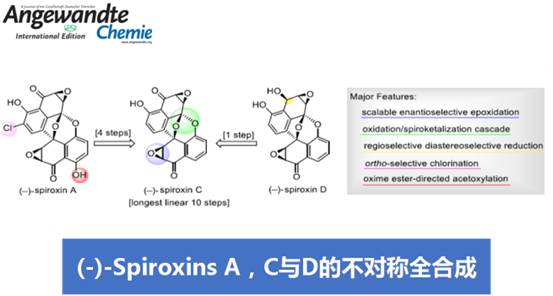
Spiroxins A、C与D是在海洋真菌菌株 (marine fungal strain) LL-37H248中分离鉴定出的代谢产物,因其具有独特的多环结构,而备受合成化学家的广泛关注。近日,西北大学胡向东课题组在Angew. Chem. Int. Ed.中发表论文,报道一种基于5-取代萘醌的克级规模对映选择性环氧化、氧化/螺缩酮化串联反应 (oxidation/spiroketalization)、苯酚结构单元的邻位选择性氯化以及肟酯导向的乙酰氧基化的关键反应步骤,进而实现(-)-Spiroxins A与C的对映选择性全合成以及(-)-Spiroxins D的首次全合成。
Enantioselective Total Synthesis of (-)-Spiroxins A, C and D
X. Shu, C. Chen, T. Yu, J. Yang, X. Hu, Angew. Chem. Int. Ed. 2021, 60, 18514. doi: 10.1002/anie.202105921.

正文
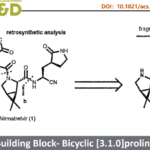
海洋真菌 (Marine fungi)已经成为具有分子结构多样化的相关代谢产物的重要来源。1999年,McDonald等[1]首次报道在真菌菌株LL-37H248中,分离出相应的天然产物Spiroxins A-E。值得注意的是,Spiroxin A在具有优良的抗肿瘤活性与抗菌活性。在分子结构中,Spiroxins A-E具有共同的两重萘醌环氧化物通过C-C键以及螺环缩酮单元结合形成的多环骨架结构I (Figure 1)。同时,这一扭曲的类笼状骨架 (distorted cage-like frame)中具有六或七重的立体中心。
2003年,Imanishi等[2]首次采用TBAF活的化Suzuki-Miyaura交叉偶联反应作为关键步骤,首次实现(±)-Spiroxin C的全合成。2017年,基于萘醌衍生物立体专一性分子内光氧化还原反应策略,Suzuki等[3]首次完成(-)-Spiroxin C的对映选择性全合成。之后,Suzuki等[4]报道采用光辐射或酸/碱条件引发的两种不同的串联反应方法学策略,分别使相同中间体的绝对构型发生保持与翻转。同时,Suzuki团队通过上述策略作为关键步骤,首次完成Spiroxin A的两种对映体的全合成。
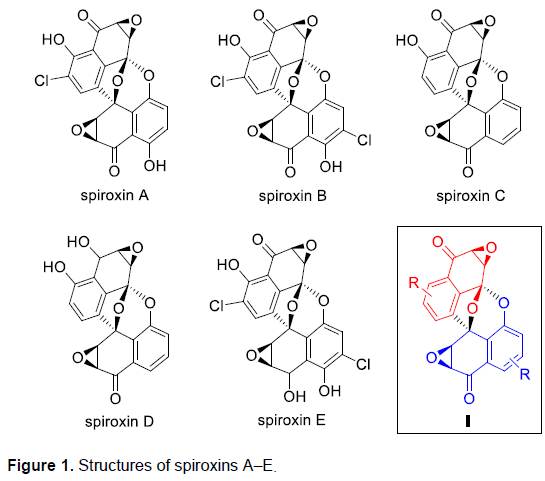
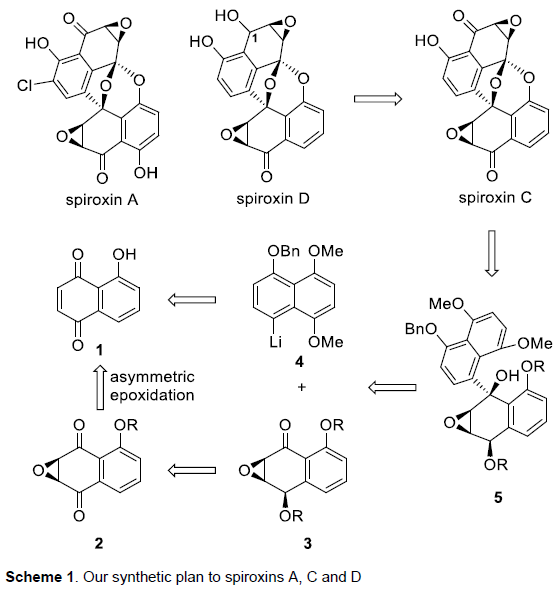
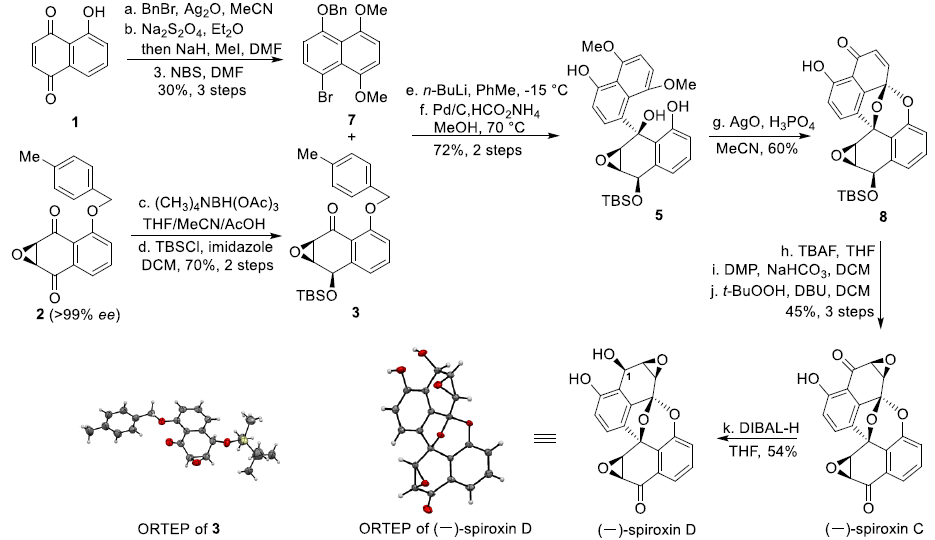
Spiroxin D与Spiroxin C分子中存在的唯一差别在于C1位的羟基 (其相对构型未知),而Spiroxin A与Spiroxin C的差别则在于,Spiroxin A分子中具有多重的羟基官能团,并存在氯取代基。由此,作者设想,Spiroxin C可能作为合成Spiroxin A与D的共同中间体。同时,对于核心多环骨架的构建,作者设想通过5的氧化以及后续螺缩酮化的一步串联反应过程而实现。其中,通过选择锂试剂4与手性α,β-环氧酮3之间的非对映选择性加成策略,完成5的制备。而3与4均可由商品化的juglone 1进行制备,并能够采用对映选择性环氧化过程,在2分子中引入初始的手性立体生成中心,进而能够成功实现Spiroxins A、C以及D的对映选择性全合成。
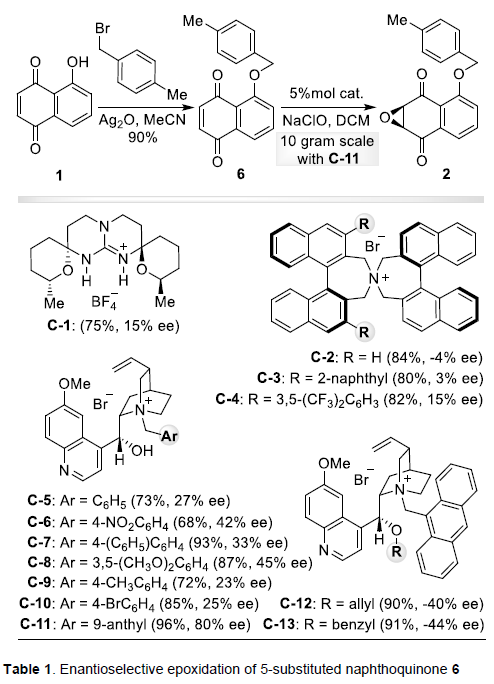
该小组面临的首要挑战在于实现手性α,β-环氧酮3的高度对映选择性合成。作者首先通过市售的juglone 1,完成化合物6的制备。之后,作者进一步对5-取代萘醌6的对映选择性环氧化反应中的不对称相转移催化剂进行筛选 (Table 1, C-1至C-13)。研究表明,采用C-11时,在相应的克级规模实验中,能够获得96%收率的环氧化产物2,ee为80%。之后,作者观察到,通过重结晶过程,能够实现有效的对映体富集 (>99% ee)。
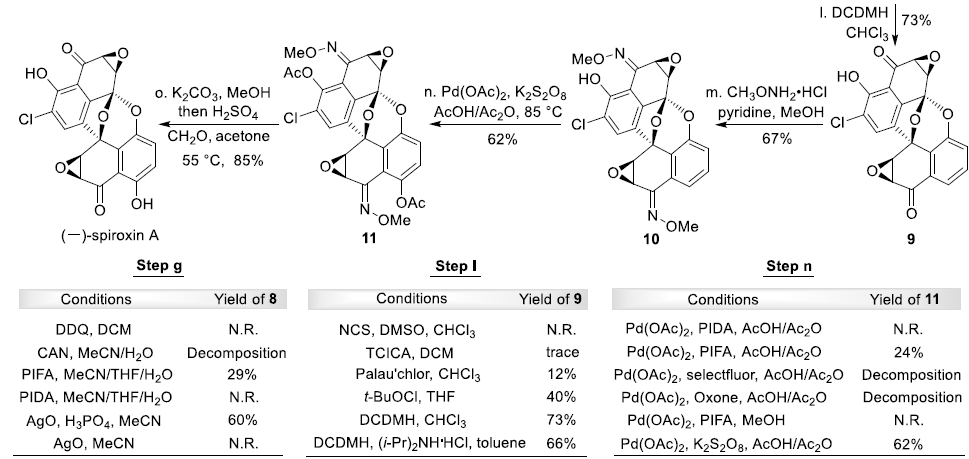
接下来,作者开始进行(-)-Spiroxin C的全合成方案设计 (Scheme 2)。首先,作者对手性环氧化物2相关的还原反应条件进行深入研究。进而观察到,在采用(CH3)4NBH(OAc)3作为还原剂时,能够获得优良的区域与非对映选择性,并通过后续的羟基保护步骤,形成产物3。X-射线晶体学研究表明,产物3中环氧化物片段的绝对构型与(-)-Spiroxins A以及C中的环氧化物片段相同。同时,juglone 1经苄基保护、醌还原-二甲基化以及溴化的三步反应过程,形成芳基溴产物7。之后,环氧化物3与通过芳基溴7原位形成的芳基锂试剂作用,并经历进一步的苄基与4-甲基苄基去除步骤之后,形成砌块5。接下来,该小组设想,通过螺缩酮单元的引入,进而顺利完成分子中核心多环骨架的构建。这里,作者发现,在采用氧化银(II)与磷酸氧化体系[5]时,能够成功获得60%收率的螺缩酮产物8。其中,磷酸的存在对于氧化/螺缩酮化串联反应过程的顺利进行尤为关键 (参阅Supporting Information)。之后,通过后续的去除TBS保护基团、羟基官能团的氧化以及环氧化步骤,进而顺利完成(-)-Spiroxin C分子的全合成。接下来,作者进行C1构型未知的 (–)-spiroxin D的全合成研究。实验发现,在采用DIBAL-H还原(–)-Spiroxin C中的C1羰基时,能够直接获得(–)-Spiroxin D,并表现出优良的区域选择性与非对映选择性。同时,通过上述转化过程,并进一步结合X-射线晶体学分析,进而顺利确定 (–)-spiroxin D分子中C1位置的绝对构型。
最后,为完成(-)-Spiroxin A的全合成,作者首先对 (-)-SpiroxinC分子中苯酚片段的邻位选择性氯化过程进行深入研究。实验表明,在采用1,3-二氯-5,5-二甲基乙内酰脲 (1,3-dichloro-5,5-dimethylhydantoin, DCDMH)作为氯化试剂时,能够获得73%收率的氯化产物9。同时,(-)-Spiroxin A的全合成设计中面临的巨大挑战,则在于区域选择性地将羟基引入至(-)-Spiroxin A分子中最下端的芳环片段。其中,作者设想,采用肟酯导向的乙酰氧基化反应策略[6]将能够有效地解决上述问题。并且,作者发现,选择K2S2O8作为氧化剂时,能够获得62%收率的关键砌块11。接下来,通过去除分子中全部的乙酰基保护基与肟酯基团之后,最终完成(-)-Spiroxin A的全合成。

总结
西北大学胡向东课题组分别通过10、11以及14步的反应设计方案,顺利完成(-)-Spiroxins C、D与A的对映选择性全合成。其中,关键的反应步骤涉及5-取代萘醌的对映选择性环氧化、氧化/螺缩酮化串联反应、通过DCDMH参与的苯酚片段的邻位选择性氯化以及肟酯导向的乙酰氧基化过程。















No comments yet.