
本文作者:杉杉
导读
电化学在有机合成中具有诸多优势,而且,在电化学条件下能够实现一系列不同类型的氧化还原反应。然而,涉及电化学条件下进行的C-C键断裂与官能团化的反应方法学研究则较少有文献报道。近日,武汉大学雷爱文教授课题组开发出一种无催化剂 (catalyst-free)与无外加氧化剂 (external-oxidant-free)存在的电化学反应策略,进而成功实现芳基环丙烷C-C 键的断裂与1,3-双官能团化。同时,作者发现,通过选择不同类型的亲核试剂,能够进一步实现芳基环丙烷底物的1,3-二氟化 (1,3-difluorination)、1,3-氧氟化(1,3-oxyfluorination)与1,3-双氧化 (1,3-dioxygenation)反应,并获得高度的化学与区域选择性。该方法学具有良好的官能团兼容性,并且,在克级规模的实验条件下,同样能够有效地进行。同时,反应机理研究表明,通过阳极氧化产生的芳基环丙烷自由基阳离子与随后产生的苄基碳正离子为反应过程的关键中间体。
Electrochemical C-C bond cleavage of cyclopropanes towards the synthesis of 1,3-difunctionalized molecules
P. Peng, X.Yan, K. Zhang, Z. Liu, L. Zeng, Y. Chen, H. Zhang, A. Lei,
Nat. Commun. ASAP. doi: 10.1038/s41467-021-23401-8.
正文
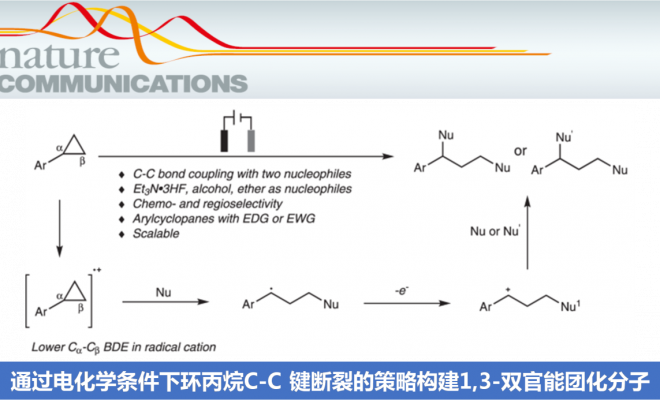
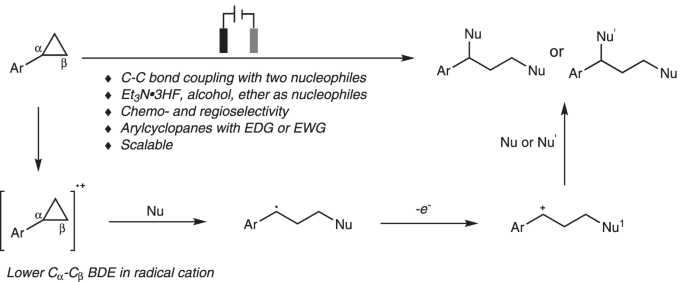
C-C键是构成有机分子骨架的基本成键单元。并且,通过C-C键的断裂,能够直接实现一系列不同类型的官能团化反应方法学,进而更加高效地完成各类复杂有机分子的构建。环丙烷作为重要的合成砌块,通过环丙烷分子中环张力的释放,进而促进其顺利开环的反应策略,已经广泛应用于各类复杂天然产物的全合成。其中,对于各类DAC (donor-acceptor cyclopropane)底物而言,由于存在固有的电子偏倚 (electronic bias),因此,能够采用Lewis酸催化剂,有效地促进各类DAC分子的开环[1-3]。而对于非活化的环丙烷,目前,已经发展出两种不同方式的开环策略,即采用过渡金属试剂参与的氧化加成[4] (这种开环方式仅限于开环重排或环加成反应,并且需要引入特定的导向基团,进而实现其区域选择性的开环官能团化)与通过Lewis酸配合物促进的亲电活化反应[5-13] (这一策略仅限于亲电加成反应)。同时,在上世纪70年代 [14-15],已经发展出通过芳基环丙烷分子经历单电子氧化过程产生的自由基阳离子引发的开环官能团化策略。近年来,这一策略已经进一步应用于在氧化剂或光存在下进行的芳基环丙烷底物的 1,3-氨基官能团化 (1,3-aminofunctionalization)、1,3-氧胺化 (1,3-oxoamination)与1,3-氧氯化 (1,3-oxochlorination)反应。然而,在多数情况下,上述策略并未涉及克级规模反应的研究。并且,各类带有吸电子基团取代的芳基环丙烷 (具有较高的氧化电势)无法与上述策略良好的地兼容 (Fig. 1a) [16-20]。
有机电化学策略由于具有易于进行工业大规模生产、无需化学计量的氧化剂或还原剂研究良好的反应可调控性 (reaction tunability)等优势,在有机合成方法学的研究中逐渐兴起[21]。通过这一策略,能够实现各种不同类型的氧化还原反应[22-34]。 然而,由于 C-C键的惰性以及其较弱的电子偏倚,因此,采用电化学氧化促进的C-C键断裂/官能团化方法学极少有文献报道[4], [35-36]。1970年,Shono团队首次报道芳基环丙烷在甲醇媒介中进行的阳极氧化的过程[37]。然而,仅获得六种相关的目标产物 (Fig. 1b)。受上述报道的启发,作者设想 (Fig. 1c),将芳基环丙烷通过阳极氧化,形成自由基阳离子,并进一步通过三电子SN2反应,产生苄基自由基,之后,苄基自由基继续在阳极失去电子,转化为苄基碳正离子。接下来,通过不同亲核试剂对苄基碳正离子的进攻,最终获得一系列1,3-双官能化产物。
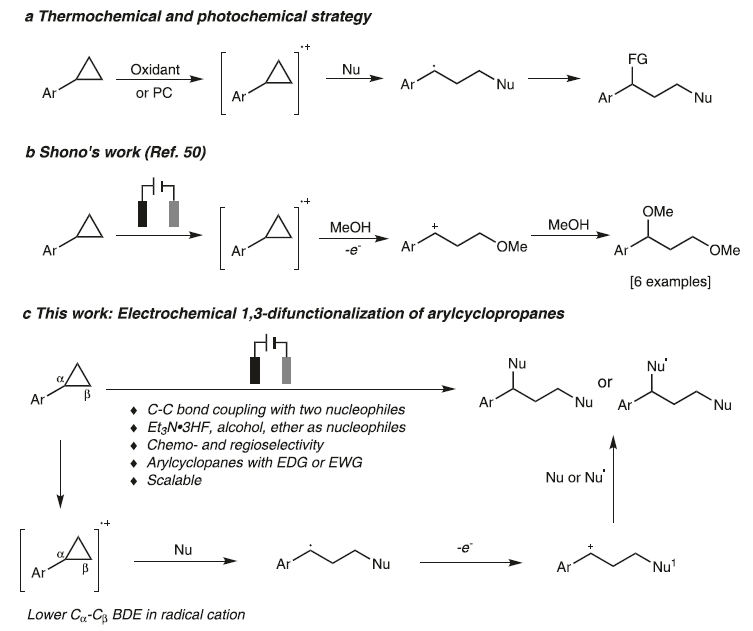
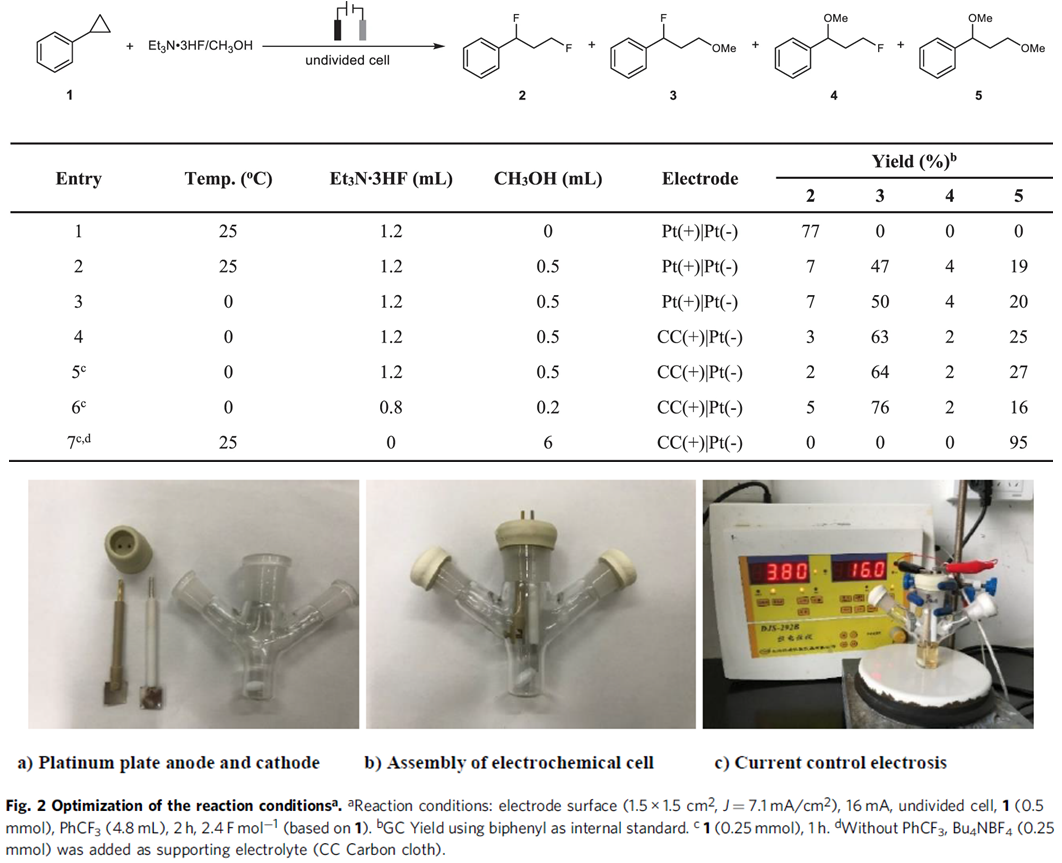
基于上述设想,首先,作者采用苯基环丙烷 1作为模型底物,进行了相关反应条件的优化筛选 (Fig. 2)。研究表明,选择Et3N·3HF作为氟源 (fluorine source),铂板 (platinum plate)作为阳极与阴极,在无隔膜电解槽 (undivided cell)中,反应温度为25 oC, 采用16 mA的恒定电流 (constant current)下进行电解,最终获得77% 收率的1,3-二氟化产物 (1,3-difluorination) 2。同时,作者发现,将反应温度降至0 oC,改用碳布 (carbon cloth)作为阳极,并进一步调整Et3N·3HF/MeOH的体积比,能够获得76%收率的1,3-氧氟化 (1,3-oxyfluorination)产物 3。而采用 Bu4NBF4作为支持电解质 (supporting electrolyte),碳布作为阳极,反应温度为25 oC,无相应氟源存在的条件下进行电解,则可以获得 95%收率的1,3-双氧化(1,3-dioxygenation)产物 5。
在获得上述最佳反应条件后,作者对1,3-二氟化反应 (1,3-difluorination reaction)的底物适用范围进行考察 (Fig. 3)。研究表明,一系列带有吸电子基团取代的芳基环丙烷,均能够顺利完成相应的电解转化过程,并以中等至良好的收率获得1,3-二氟化产物 2–22以及25。然而,除TMS基团 (获得产物 23)与烷基 (获得产物 24)外,其它各类带有供电子基团取代的芳基环丙烷底物,均无法与上述最佳反应条件有效地兼容。同时,实验观察到,杂芳基环丙烷底物,例如噻吩基环丙烷,同样能够有效地完成上述电解反应,并获得产物 26。此外,实验发现,1,1-二取代环丙烷、1,2-二取代环丙烷以及三取代环丙烷底物,同样能够顺利进行上述1,3-二氟化反应,获得相应目标产物 27–31。而且,作者进一步发现,上述的1,3-二氟化反应方法学同样能够成功应用于一系列复杂天然产物后期的衍生化过程,并获得相应衍生化产物 32–35。最后,通过对产物6、10、14与22的克级实验,进一步证实上述1,3-二氟化反应方法学的合成应用价值。
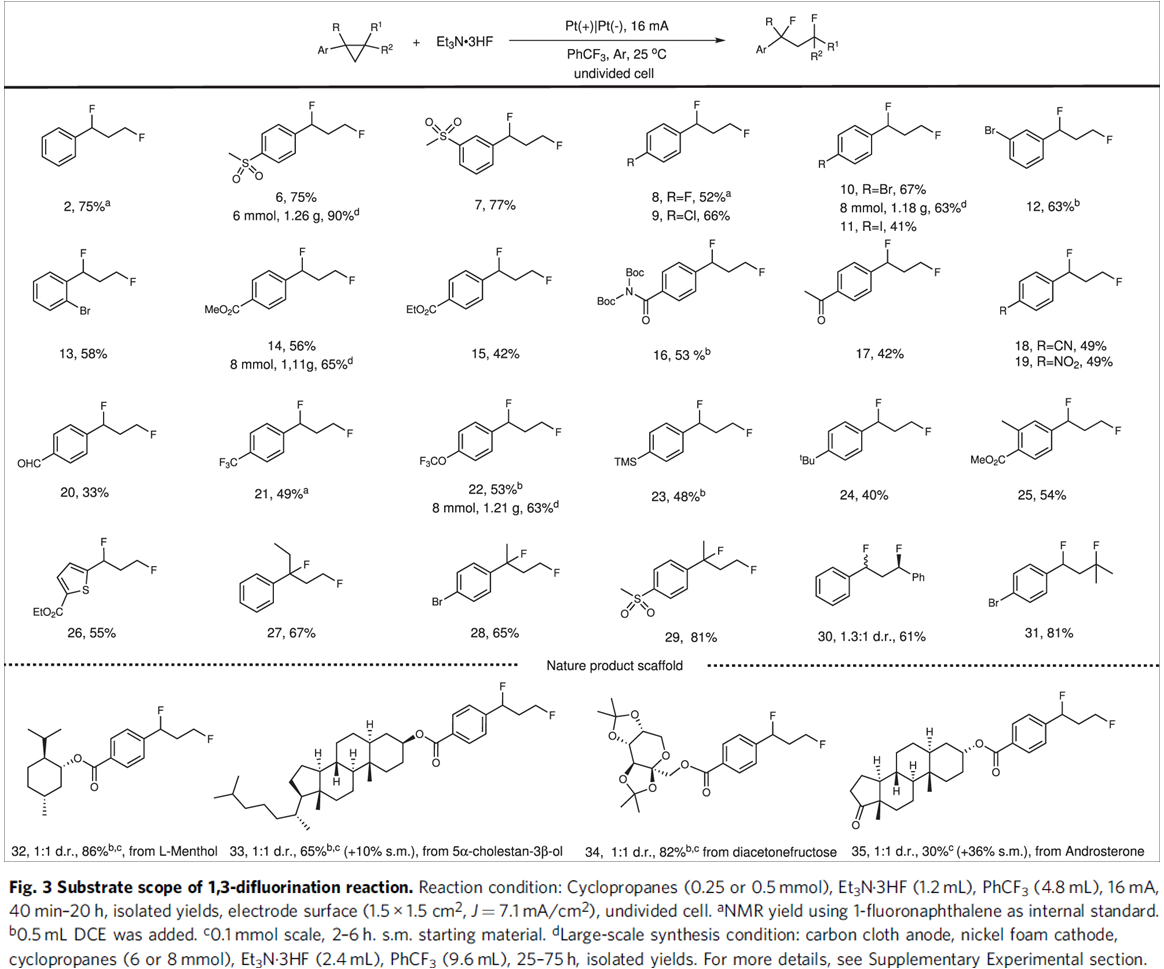
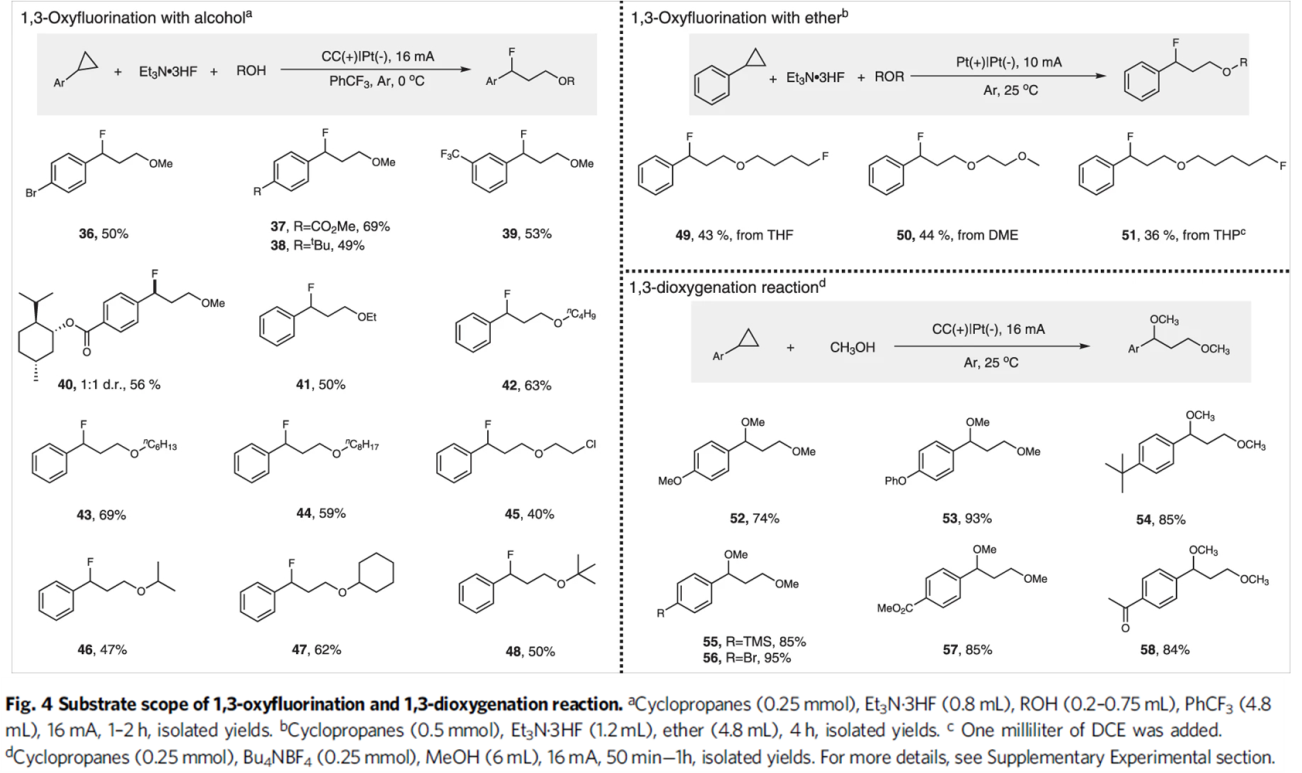
接下来,作者进一步考察1,3-氧氟化反应 (1,3-oxyfluorination reaction)的底物应用范围 (Fig. 4)。该小组发现,一系列带有供电子基与吸电子基取代的芳基环丙烷底物,均能够有效地与上述电解反应的条件兼容,并以中等程度的反应收率获得相应1,3-氧氟化产物36–40。同时,该小组对醇底物的适用范围进行考察。实验结果表明,除甲醇外,EtOH与n-BuOH以及长链一级醇均能够有效地参与相应的1,3-氧氟化反应,并获得产物41–44。2-氯-1-乙醇同样能够以40%的收率获得产物45。并且,二级醇与三级醇同样能够与上述最佳的电解反应条件良好地兼容,并获得产物46–48。值得注意的是,除醇类化合物之外,醚类化合物同样能够顺利参与上述1,3-氧氟化反应,并获得产物49–51。最后,作者进一步对1,3-双氧化反应的底物应用范围进行研究。实验观察到,一系列带有供电子基以及吸电子基取代的芳基环均能够与上述最佳的电解反应的条件良好地兼容,并能够以良好至优良的反应收率获得相应的产物52–58。
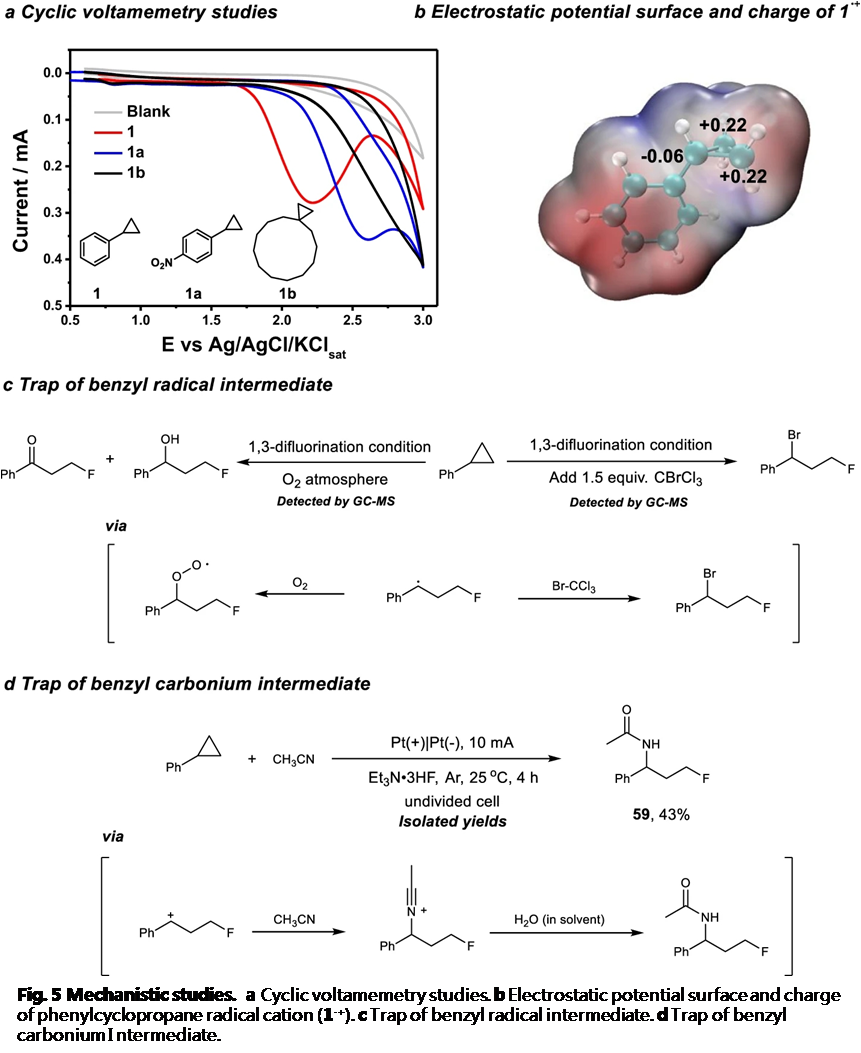
为阐明合理的反应机理,作者进行一系列相关的实验研究 (Fig. 5)。首先,通过循环伏安法 (cyclic voltammetry)实验,测定环丙烷底物的氧化还原电势 (redox potential)。研究发现,烷基取代的环丙烷1c具有较高的氧化电势,并且,在3V的电压下,未观察到氧化峰 (oxidation peak) (Fig. 5a)。相比之下,具有强吸电子基团取代的芳基环丙烷 (1a)则具有相对较低的氧化电势。上述事实表明,芳基取代基对于环丙烷底物的氧化起关键作用。同时,作者通过 DFT 计算进一步研究由苯基环丙烷氧化形成的相应自由基阳离子的电荷分布。计算表明,环丙烷远端的碳原子(distal C atom)带有部分正电荷,是潜在的亲核进攻位点 (Fig. 5b)。此外,由于苄基自由基与氧气之间能够十分迅速地进行反应,因此,作者将上述反应在氧气气氛中进行,从而捕获可能的苄基自由基中间体。研究表明,反应过程中,苄基自由基的形成,对于氧化产物 (oxygenation product)的检测表现出高度的信号感应。同时,采用 BrCCl3进行的自由基捕获实验 (trapping experiment),同样能够进一步证实苄基自由基的存在 (Fig. 5c)。最后,该小组发现,将苯基环丙烷与Et3N·3HF在CH3CN 溶剂存在的条件下进行电解反应,最终能够分离出酰胺化产物59,从而表明反应过程中涉及苄基碳正离子中间体的产生 (Fig. 5d)。
总结
武汉大学雷爱文教授课题组成功开发出通过芳基环丙烷的电化学氧化 C-C键断裂的策略,能够顺利完成各类芳基环丙烷底物的1,3-双官能团化反应,从而获得一系列1,3-二氟化、1,3-氧氟化与1,3-双氧化产物。反应过程中,无需催化剂与外加氧化剂。作者进一步研究表明,上述电化学氧化策略具有良好的官能团兼容性,并且,能够有效地实现相应的克级规模反应。相关的机理研究表明,阳极氧化中产生的芳基环丙烷自由基阳离子与后续形成的苄基碳正离子为反应过程的关键中间体。













No comments yet.