

译自Chem-Station网站日本版
原文链接:アレ?アレノン使えばノンラセミ化
本文翻译:asymmboy
校对: JoJo
本文作者报道了一种全新的无外消旋化的多肽合成策略。这一策略采用联烯酮 (allenone)作为缩合试剂,能够以较高的反应收率获得相应的二肽产物。同时,这一全新的酰胺偶联策略同样能够用于液相多肽片段连接与固相多肽合成。
肽缩合剂的发展
自1901年Fischer小组完成首例二肽分子合成之后,多肽合成已历经100多年的历史。随着多肽在药物研发与材料化学领域中 (精细化学品研究中的核心领域)的需求日益增加,多肽合成策略的研究已经取得诸多飞跃性的进展。然而,目前最为通用的多肽合成策略仍然是采用缩合试剂与天然α-氨基酸形成活化酯中间体,进而与另外一分子α-氨基酸通过氨解反应,实现肽键的构建。迄今为止,有机合成化学家已经成功开发出多种不同类型的酰胺偶联缩合试剂。
首例获得广泛应用的缩合试剂为Sheehan小组开发的二环己基碳二亚胺(dicyclohexylcarbodiimide, DCC)试剂 (Figure 1A) [1]。之后,Stevens与Munk小组选用烯酮亚胺 (ketenimine)作为缩合试剂,顺利完成相应肽键的构建 (Figure 1B) [2]。然而,在上述偶联策略中,由于这类缩合试剂结构中带有碱性位点,会诱发通过噁唑酮 (oxazolone)中间体形成 (Path A)或通过羰基α-位的直接分子内质子攫取 (Path B),而使形成的肽或酰胺产物出现显著的外消旋化[3]。为抑制外消旋化的发生,由DCC缩合剂参与的反应中,需要外加HOBt (1-hydroxybenzotriazole)、HOAt (1-hydroxy-7-azabenzotriazole)以及Oxyma (ethyl 2-cyano-2-(hydroxyimino)acetate)等消旋抑制剂,通过现场生成形成的活化酯,以避免外消旋化。
近期赵军锋课题组报道了一种炔酰胺 (ynamide)缩合试剂介导的无外消旋化(racemization-free)的多肽合成方法学 (Figure 1C)[4]。这一策略中,炔酰胺氮原子中的吸电子基团Ts发挥着重要的作用,它使炔酰胺缩合试剂的碱性显著降低,进而有效地避免了目标产物的外消旋化。然而,由于炔酰胺试剂的反应活性过于温和,因此不利于其固相肽合成中的应用。
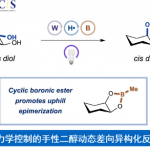
之后,赵军锋课题组发现DCC、烯酮亚胺与炔酰胺缩合试剂分子结构中均含带有一个亲电性的sp碳原子,其对于活化酯的形成尤为重要。另外,该小组还注意到,活化酯结构中的碱性位点是导致产物外消旋化的关键因素。因此,该小组提出,由同样具有亲电性sp碳原子的联烯衍生物中,发现一种不含碱性位点的缩合试剂的研究设想,并最终成功开发出全新的联烯酮缩合试剂。联烯酮通过1,4-加成/异构化串联反应与α-氨基酸形成反应活性适中的α-羰基乙烯酯中间体 (α-carbonyl vinyl ester intermediate),从而顺利解决多肽合成过程中,α-手性中心外消旋化的问题 (Figure 1D)。

Figure 1. (A) Sheehan等发展的反应策略 (B) Stevens等发展的反应策略 (C) Zhao等发展的反应策略 (D) 本文中作者发展的反应策略
Allenone-Mediated Racemization/Epimerization-Free Peptide Bond Formation and Its Application in Peptide Synthesis,
Z. Wang, X. Wang, P.Wang, J. Zhao, J. Am. Chem. Soc. 2021, 143, 10374. doi: 10.1021/jacs.1c04614.
论文作者简介

论文作者: Junfeng Zhao, 赵军锋 (Symform, 2019, PDF)
科研经历:
1998-2001 B.S., Beijing Normal University
2002-2005 M.S., Central China Normal University (Prof. Mingwu Ding)
2005-2006 Ph. D. candidate, Chengdu Institute of Organic Chemistry (Prof. Liuzhu Gong)
2006-2010 Ph. D. Nanyang Technological University (Prof. Teckpeng Loh)
2010-2011 Postdoc, Nanyang Technological University (Prof. Chuanfa Liu)
2011-2013 Postdoc, University of Bonn (Prof. Michael Famulok) and University of Münster (Prof. Armido Studer)
2013-2014 Assistant Professor, University of Hong Kong (Prof. Dan Yang)
2014- Professor, Jiangxi Normal University
论文概要
研究内容:具有生物活性的多肽与蛋白质的合成以及修饰方法学研究
赵军锋课题组发展的新策略是通过α-羰基乙烯活化酯中间体的形成以及其后续氨解的两步反应,以实现酰胺键的构建 (Figure 2A)。首先,羧酸1与联烯酮2在二氯乙烷溶剂中通过1,4-加成/异构化串联反应,以良好的收率获得α-羰基乙烯酯中间体3。该反应具有良好的底物适用范围,脂肪族羧酸 (1a)、芳香族羧酸(1b)以及α及族-不饱和羧酸 (1c 与1d)底物均能够有效地兼容,并以良好的反应收率获得相应的α-羰基乙烯酯产物3a–3d (Figure 2B)。之后,作者在DMF溶剂中,将α-羰基乙烯酯3与一系列胺底物4进行进一步的氨解反应,最终获得相应的酰胺产物5。具有较高立体位阻的胺底物 (4a)以及亲核性较弱的芳胺底物(4d)同样能够良好地兼容,尽管其酰胺化反应速率较慢,然而,作者发现,催化量的HOBt(10%)能够显著加快氨解的反应速度,并以优良的反应收率,获得酰胺产物5a与5d。同时,作者进一步发现,α-羰基乙烯酯3中间体具有极高的生成效率,并具有极高的反应活性。因此,上述两步反应可以在“一锅”条件下进行,反应收率无明显降低。接下来,作者对这一全新的酰胺偶联方法学在肽键形成中的适用性进行考察。研究表明,天然与非天然氨基酸底物均能够以较高的反应收率获得相应的二肽产物,值得注意的是,在相关的反应过程中,均未观察到α-手性中心的外消旋化 (Figure 2C)。具有较高立体位阻的N-甲基氨基酸 (5e)、具有游离羟基的苏氨酸 (threonine)与丝氨酸底物 (serine) (5f 与 5g)以及吲哚NH未保护的色氨酸 (tryptophan) (5h)等底物,均能够高效地参与相应二肽分子的合成。此外,上述策略同样能够应用于液相多肽片段连接与固相多肽合成。综上表明,这一全新的酰胺偶联策略具有极佳的合成实用性。
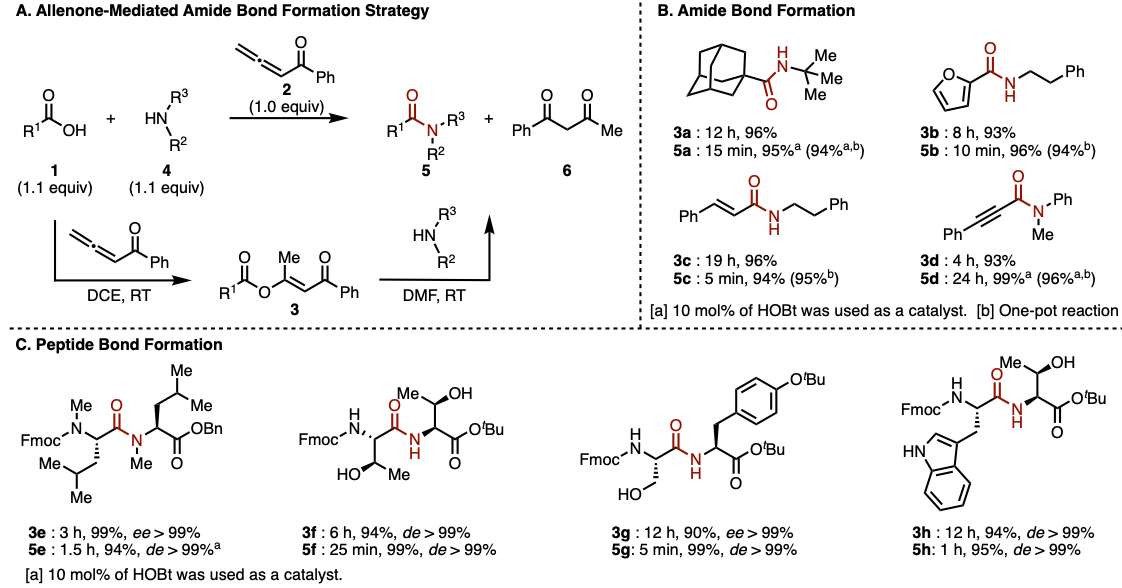
Figure2. (A) 反应路径 (B)酰胺键的形成 (C) 肽键的形成
综上所述,赵军锋课题组成功开发了一种采用联烯酮作为偶联试剂的肽键形成方法。该策略成功解决了多肽合成中最为棘手的外消旋化或差向异构化问题。因此,这一全新的偶联策略将来有望在与酰胺以及多肽合成相关的研究领域中作出巨大的贡献。
参考文献
- C.Sheehan, G. P. Hess, J. Am. Chem. Soc. 1955, 77, 1067. doi: 10.1021/ja01609a099.
- L.Stevens, M. E. Munk, J. Am. Chem. Soc. 1958, 80, 4069. doi: 10.1021/ja01548a060.
- El-Faham, F. Albericio, Chem. Rev. 2011, 111, 6557. doi: 10.1021/cr100048w.
- Hu, S. Xu, Z. Zhao, Y. Yang, Z. Peng, Yang, M. Wang,J. Zhao, J. Am. Chem. Soc.2016, 138, 13135. doi: 10.1021/jacs.6b07230


















No comments yet.