
本文作者:杉杉
导读:
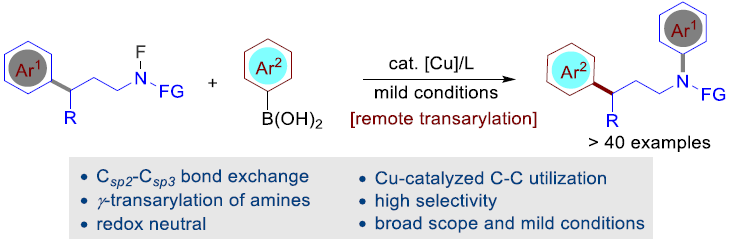
通过C-C键官能团过程进行的胺分子的远程芳基化,目前尚未有文献报道。近日,南方科技大学舒伟课题组在ACS Catal. 发表论文,报道了在温和的铜催化条件下,通过非张力C-C键的断裂,从而实现胺的γ-迁移芳基化(γ-transarylation)。同时,对于机理的研究表明,这种氧化还原中性反应(redox-neutral reaction)经历阻断的自由基途径(intercepted radical pathway),并涉及Csp2-Csp3键断裂。之后,再与铜催化的芳基化反应(在芳基硼酸存在下)结合,从而构建新的Csp2-Csp3键。
Cu-Catalyzed Remote Transarylation of Amines via Unstrained C-C Functionalization
Y. Wang, J. Zhang, W. Shu, ACS Catal. ASAP. DOI:10.1021/acscatal.0c0471
正文
脂肪胺广泛存在于各类药物分子与天然产物中,在当前最畅销的200种药物分子中,多数分子中存在脂肪胺骨架。因而,对于有机与药物化学家而言,开发一些简单有效的脂肪胺分子修饰与转化的方法学极尤为重要。而脂肪族胺的位点选择性芳基化反应,由于能够避免起始原料的前期官能团化(prefunctionalization)步骤,因而,通过惰性键官能团化(如C-H或C-C键官能团化)的方法学,在所有具有潜在应用价值的方法中极具吸引力。在过去十年中,胺的γ-选择性C-H芳基化已经取得较多的研究进展(Figure 1a)。Sanford等[1]通过开发出通过胺C-H键的断裂,从而实现其构象导向(conformation-directed)的γ-芳基化反应方法学。Yu, Gaunt等[2]报道了过渡金属催化(导向基团促进)胺的γ-C-H芳基化反应。最近,Yu,Dong与Ge等[3]报到了采用瞬态导向基团(transient directing group)实现Pd催化胺的γ-C-H芳基化反应。相比之下,由于饱和C-C键的惰性与立体位阻以及同一分子中存在多种C-H键,因此,至今尚未开发出能够通过C-C键活化的方式对其分子骨架进行有效修饰的方法。并且,通过C-C键官能团化进行的胺分子γ-芳基化同样无相关的文献报道(Figure 1b)。由此,作者设想,发展一种涉及由氮中心自由基引发的芳基迁移/铜催化交叉偶联反应,从而能够在温和的条件下,通过C-C键的断裂,实现胺的γ-芳基化反应,。另一方面,自由基引发C-C键断裂是有机化学新兴的研究领域。尤其是芳基迁移反应已经成为C-C键活化并形成新的碳、氮以及氧中心自由基的一种有效工具。近期,Shi,Zhu与Bi等[4]报道了在氧化条件下,通过自由基途径的(杂)芳基的迁移,实现C-C键断裂的方法学。然而,迄今为止,将通过非张力C-C键断裂产生的自由基捕获,并进一步参与金属催化的交叉偶联反应,由于反应条件较为苛刻,并且,产生的中间体与金属催化剂难以兼容。目前,文献中尚未有成功的反应报道。这里,南方科技大学的舒伟课题组报道了在铜催化条件下,通过将C-C键的断裂与铜催化交叉偶联反应结合,从而实现胺分子的γ-迁移芳基化反应(Figure 1c)。作者观察到,在铜催化的条件下,能够以高度选择性的方式活化C-C键。此外,选用带有N-F键的胺底物,在氧化还原中性条件下,能够将Csp2-Csp3键的断裂与Csp2-Csp3键的形成有效地结合。与此同时,Cook、Zhu、Nagib、Li与Muñiz等[5]在近期报道了基于铜氧化还原催化的N-F键活化与合成应用策略。该策略中,采用预先形成的N-F键通过1,n-HAT(n = 5或6)的方式,进而引发位点选择性的C-H官能团化。然而,这一策略中,C-C键的断裂过程并未涉及N-F的参与。

首先,作者选择1a与4-氟苯硼酸作为模型底物,进行了相关反应条件的筛选(Table 1)。确定出反应的最佳条件为:Cu(OAc)2作为催化剂,L1与L2-1作为配体,叔丁醇锂作为碱,在DCM溶剂中(含少量水),40℃下反应。

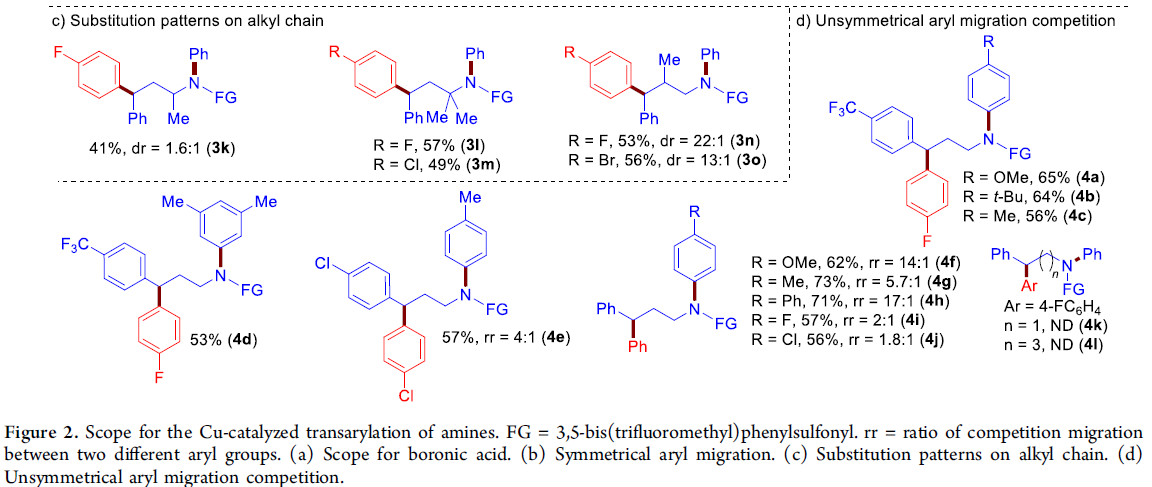
在获得上述最佳反应条件之后,作者首先对硼酸底物1的应用范围进行考察(Figure 2a)。研究表明:带有吸电子与供电子基的芳基硼酸,均能够以较高的收率获得相应的产物2a–2q。卤素取代的芳基硼酸同样能够顺利完成上述反应,获得产物2b–2d。缺电子的芳基硼酸同样能够有效地兼容(2e–2h)。同样地,具有对位、间位以及多取代的芳基硼酸同样能够有效地参与上述反应过程,并以中等至良好的收率获得相应的γ-芳基化产物2a–2q。此外,杂芳基硼酸(如苯并呋喃与噻吩基硼酸)同样能够较好地转化为γ-杂芳基胺2r与2s。
接下来,作者对胺的范围适用反应进行进一步研究(Figure 2b和2c)。作者发现,一系列芳环中带有各种富电子与缺电子取代基的γ-二芳胺,均能够顺利地进行上述反应过程,获得预期的产物3a–3j。烷基链中具有α-,α,α-或β-取代基的γ-二芳胺均能够与上述反应体系兼容,并以较高的收率与中等至优良的非对映选择性,获得相应目标产物3k–3o。
此外,作者同样对γ-二芳胺分子中不同芳基之间参与竞争性Csp2-Csp3键交换的能力进行深入考察Figure 2d)。实验结果表明,在γ-二芳胺分子中的不同芳基之间,可以观察到良好的Csp2-Csp3键交换的选择性(4a–4j)。能够以较高的收率获得富电子芳基进行迁移芳基化的产物4a–4d。即使电子效应相近的芳基取代γ-二芳胺底物,同样可以表现出显着的选择性(4e–4j)。同时,上述反应条件对于电子效应差异极小是苯基与对甲基苯基底物,同样能够在相应的迁移芳基化产物中观察到显着的选择性,即以73%的收率与5.7:1的选择性,获得产物4g。 然而,未能通过1,3/1,5-芳基迁移,获得预期的产物4k或4l。
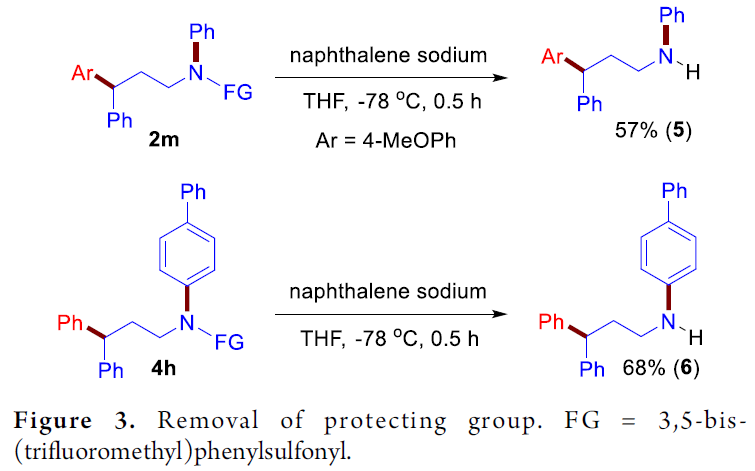
为了进一步阐明上述方法学在有机合成中潜在的应用价值,作者实验发现,将目标产物2m与4h通过萘钠处理,可以成功地去除1,3-双三氟甲基苯基磺酰基,分别以57%与68%的收率获得γ-芳基化的游离胺5与6(Figure 3)。
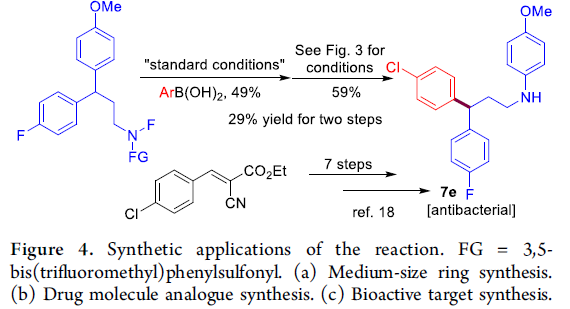
随后,作者对这一方法学的实用性进行相关的研究(Figure 4)。首先,作者将在上述的铜催化迁移芳基化方法学成功应用于扩环反应,从而合成出十元环胺7a与7b(Figure 4a)。接下来,作者将上述方法学成功应用于合成药物分子类似物Clofibrate(7c)与Beclobrate(7d)(Figure 4b)的构建。此外,该小组采用相应的N-F前体作为起始原料,运用上述迁移芳基化策略,经两步反应过程,并以29%的总收率合成出生物活性分子7e(具有潜在抗菌活性),从而简化了传统的七步合成路线(Figure 4c)。
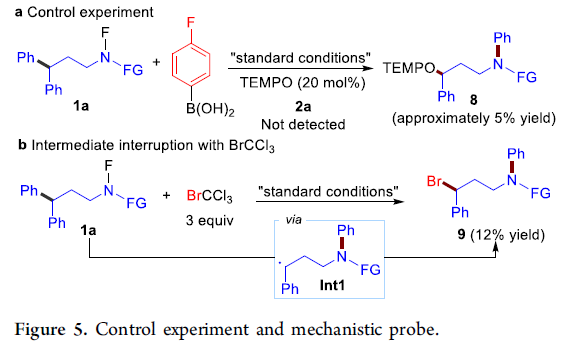
为了进一步提出反应机理,作者进行一系列相关的控制实验研究(Figure 5)。首先,作者在1a与4-氟苯硼酸的标准体系中加入20mol%的TEMPO(自由基抑制剂),发现未检测到预期的产物2a,而是仅获得5%收率的8。该结果表明,催化量的TEMPO能够完全抑制上述反应的发生。并且,通过TEMPO捕获中间体M4,最终形成8。这一实验表明,反应可能经历自由基途径(Figure 5a)。接下来,实验发现,在标准条件下,采用三氯溴甲烷代替4-氟苯硼酸时,可以获得12%分离收率的苄基溴9。该结果表明,反应过程中存在苄基自由基中间体Int1的生成,并通过溴原子转移,获得9(Figure 5b)。因此,表明该反应过程可能经历自由基途径,并涉及苄基自由基中间体Int1。
根据上述控制实验与前期的文献报道[5],作者提出一种可能的反应机理(Figure 6)。首先,在碱存在下,铜催化剂与芳基硼酸进行转金属化(transmetalation)过程,形成芳基铜配合物M1。接下来,M1通过单电子转移过程,进而使N-F键发生断裂,生成芳基铜配合物M2与氮中心自由基M3。随后,通过螺环过渡态T1进行自由基加成/芳基迁移,使M3重排,并产生碳中心自由基M4。螺环过渡态T1的稳定性影响两种不同芳基竞争性迁移的选择性。最后,M4与M2通过进一步的单电子转移过程,形成中间体Cu(III)中间体M5,再通过还原消除过程,获得最终产物并使铜催化剂再生。

总结
南方科技大学舒伟课题组报道了一种通过C-C键的官能团化实现铜催化的γ-芳基化反应方法学。同时,该方法学成功将无张力Csp2-Csp3键断裂产生的自由基与过渡金属催化的交叉偶联反应相结合。值得注意的是,底物中的N-F键具有双重作用,既可作为产生氮自由基(用于分子内芳基迁移)的前体,又可以作为有效的氧化剂,使催化剂再生,并且,使上述反应在氧化还原中性的条件下进行。




















No comments yet.