
本文作者:杉杉
导读
近日,武汉大学雷爱文教授课题组在绿色化学(Green Chemistry)发表论文,在无金属催化剂和外部氧化剂的条件下,通过电化学氧化,实现内酰胺化合物的直接C(sp3)-H杂环化反应。该反应具有广泛底物范围、温和的反应条件等优点。机理研究表明,该反应可能涉及自由基过程。此外,通过DFT计算来解释位点选择性,而克级实验进一步证明了方法的实用性。
Electrochemical Oxidative C(sp3)-H Azolation of Lactams Under Mild Conditions
Zhaohua Wan, Dan Wang, Zixuan Yang, Heng Zhang, Shengchun Wang and Aiwen Lei*
Green Chem. ASAP DOI: 10.1039/D0GC00687D
正文
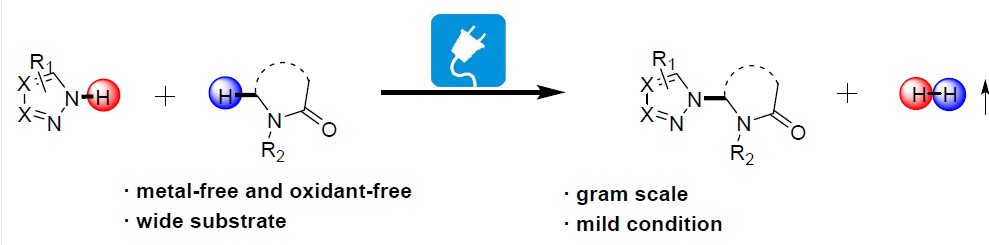
内酰胺化合物广泛存在于各类抗生素和药物中,而具有生物活性的γ-内酰胺药物最为常见。因此,内酰胺C-H功能化对此类药物的合成具有重要意义。唑化合物具有强的富电子、π–π堆积相互作用、易于形成氢键的特点,同时广泛存在于配体、离子液体和唑类药物等。基于上述讨论,若将内酰胺与唑化合物直接C-H杂环化,则可为新药的发现提供可能。近年来,已报道了使用过渡金属催化、光化学等方法实现C(sp3)-H杂环化。特别是无金属催化,但仍需要使用化学计量的氧化剂来实现这种转化,如TBHP、PhI(OAc)2、K2S2O8等。因此,仍需开发一种高效且环保的C(sp3)-H杂环化的新型体系。
氧化诱导的交叉脱氢偶联反应,作为C(sp3)-H杂环化的高效方法(Scheme 1, a)。在该反应中,与杂原子相邻的C(sp3)-H键通过单电子转移(SET)和去质子化作用生成碳正离子,再与吡咯反应,获得杂环化产物。作者设想,是否可以在电解池中,对唑进行阳极氧化以获得氮自由基,然后将其与内酰胺反应形成C-N键(Scheme 1, b)。实际上,已经报道了通过阳极氧化策略实现C-H/N-H交叉偶联的方法,并且C(sp2)-N键的形成更为常见。但是,对于C(sp3)-H的活化只有少数文献报道,这可能是由于C(sp3)-H键的化学活化能较差所致。
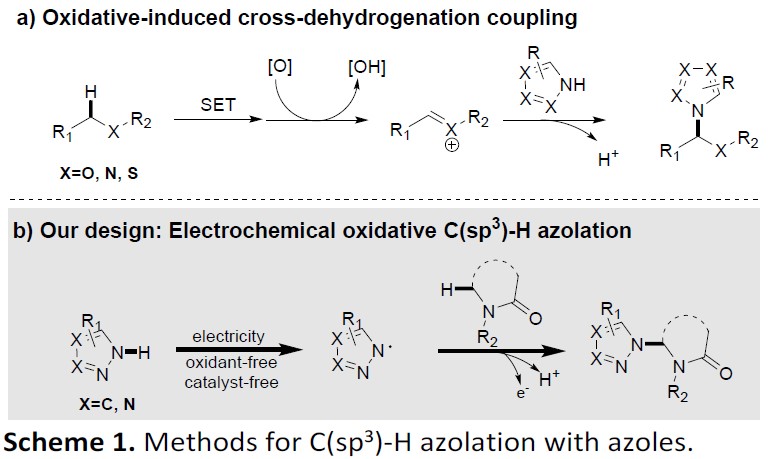
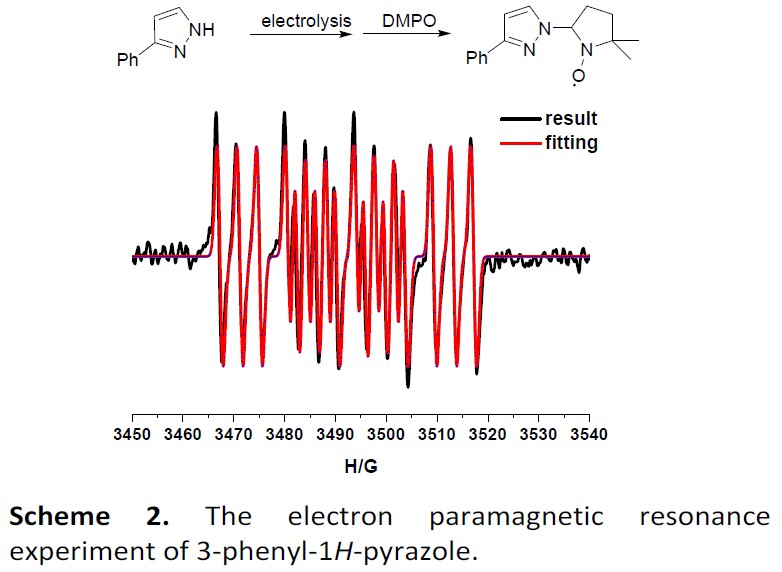
电化学氧化已成为一种高效环保的有机合成方法,同时基于本课题组长期对绿色合成方法以及各类反应机理的研究,作者通过电子顺磁共振(EPR)实验证明,在电解过程中,唑可以被氧化生成氮自由基(Scheme 2)。在此,武汉大学雷爱文教授课题组报道了,可在较温和的反应条件下,通过电化学氧化的途径,实现多种吡咯和内酰胺的直接(sp3)-H杂环化反应。
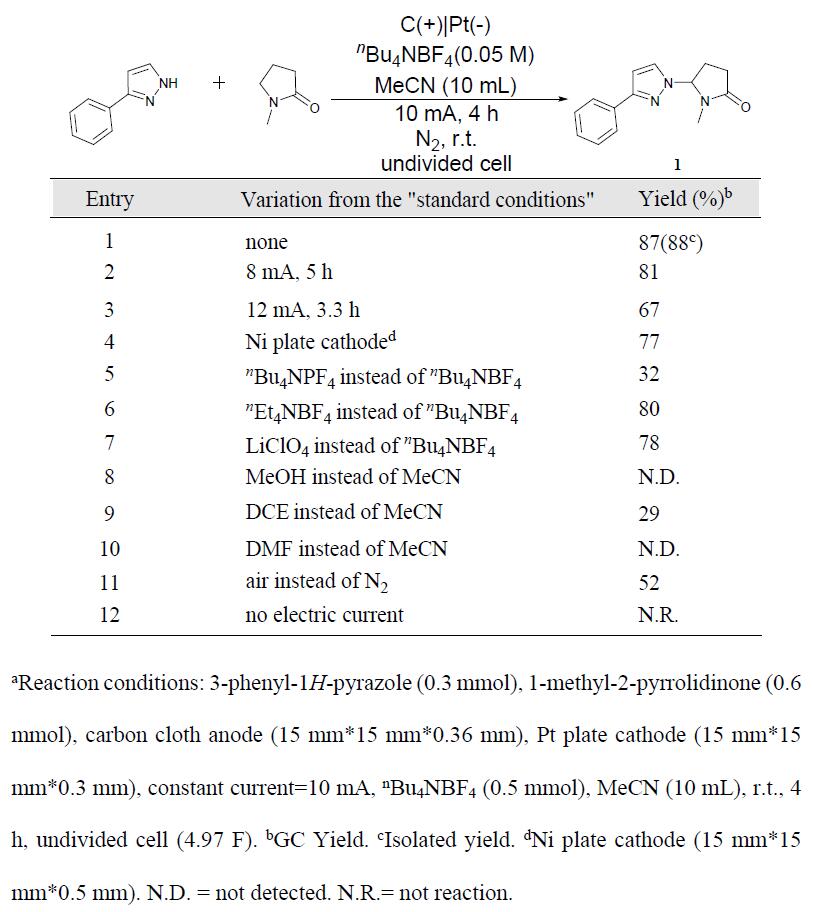
首先,作者使用3-苯基-1H-吡唑和1-甲基-2-吡咯烷酮进行反应条件的筛选(Table 2)。经过大量条件的筛选,作者发现,当使用碳阳极和铂阴极,在10 mA恒流下室温反应4 h,可获得88%的收率产物1(entry 1)。在保持电量恒定(4.97 F),适当增加或减少恒定电流,收率有所下降(entries 2-3)。而使用廉价的镍作为阴极时,收率也有所下降(entry 4)。而对电解质的筛选中,nBu4NBF4作为最佳电解质(entries 5-7)。而对溶剂的筛选时,均使收率大幅下降或不反应(entries 8-10)。此外,在空气中反应时仅获得52%收率的产物,并通过GC-MS检测到N-甲基琥珀酰亚胺(entry 11)。而在没有电流时,则不发生反应(entry 12)。
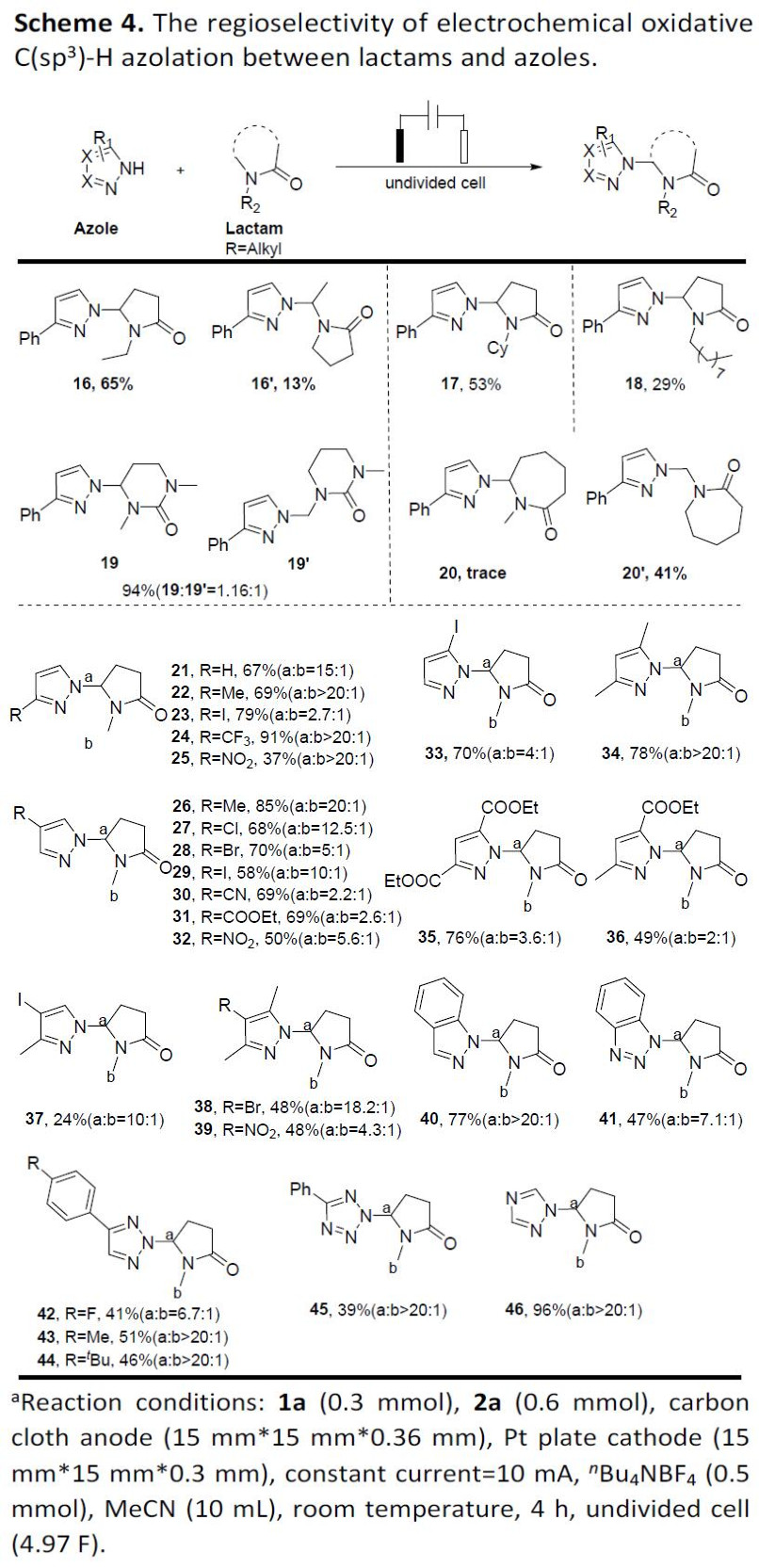
在获得上述最佳反应条件后,作者对内酰胺/酰胺的底物范围进行了研究(Scheme 3)。当使用吡咯烷-2-酮时,可获得84%收率的产物2,而六元和七元环的内酰胺,可获得中等收率的产物3、4。而带有氧原子的内酰胺,同样可以获得相应的产物5、6。但对于空间位阻较大的底物,也可获得产物7,但收率偏低。接下来,作者对酰胺作底物进行了扩展。使用N-乙酰基哌嗪可获得相应的产物8。而使用链状酰胺(如N-甲基乙酰胺、DMF、N,N-二甲基乙酰胺、N,N-二乙基丙酰胺),脲(如1,1,3,3-四甲基脲、1,1-二甲基脲、N,N’-二甲基脲)底物时,均与体系兼容,获得相应的产物9–15。

随后,作者对区域选择性进行了研究(Scheme 4)。当使用1-乙基-2-吡咯烷酮(NEP)与3-苯基-1H-吡唑反应时,异构体16和16’的比例为5:1。而吡咯烷酮中的氮原子被环己基和辛基取代时,可获得选择性产物17-18,但由于位阻原因导致收率降低。当在反应中使用脲作为环状酰胺底物时,N-甲基区域选择性19与19’的比率接近。有趣的是,N-甲基己内酰胺底物反应时,仅获得41%收率的产物20’。因此,作者发现区域选择性可能受环原子数的影响。随后,作者对多种取代唑的底物进行了区域选择性研究。当吡唑环的3,4位处被苯基、甲基、三氟甲基、硝基、卤素、氰基等取代时,获得N-CH2区域选择性偶联产物21–32(高达 20:1)。此外,吡唑的3、5位被碘原子取代,获得比例为2.7:1和 4:1的产物区域异构体产物23和33。而当吡唑的3、5位被富电子的Me基团取代时,反应以良好的区域选择性获得产物34、37、38。当引入吸电子的CO2Et(35,36),NO2(39)时,可增强了N-甲基区域选择性。此外,唑类(如吲唑、1H-苯并三唑、三唑、四唑等)也与体系兼容,获得产物40–46(收率39%-96%)。
为了进一步证明该反应的实用性,作者通过对吡拉西坦(Piracetam)和左乙拉西坦之类(Levetiracetam)的临床药物进行了修饰,并分别获得75%和55%的收率产物47和48(Scheme 5)。

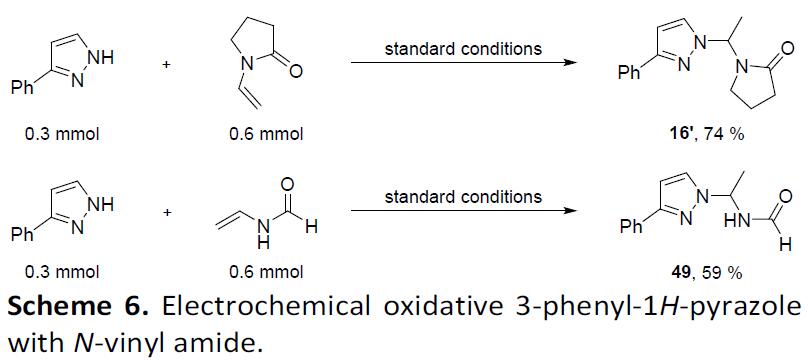
此外,作者发现,在标准条件下,使用N-乙烯基-2-吡咯烷酮与3-苯基吡唑反应时,碳的C(sp3)没有反应性,而只进行了加成反应,获得产物16’。同样,N-乙烯基甲酰胺底物也发生类似的反应,并获得碳-碳双键加成产物49,从而表明偶联产物可能涉及亲核加成过程(Scheme 6)。
为了进一步探究方法的实用性,作者进行了克级实验(Scheme 7)。当在流通池中以碳作为阳(阴)极,在60 mL的CH3CN中以20 mA恒定电流下反应33h,可获得所需产物1(收率67%)。

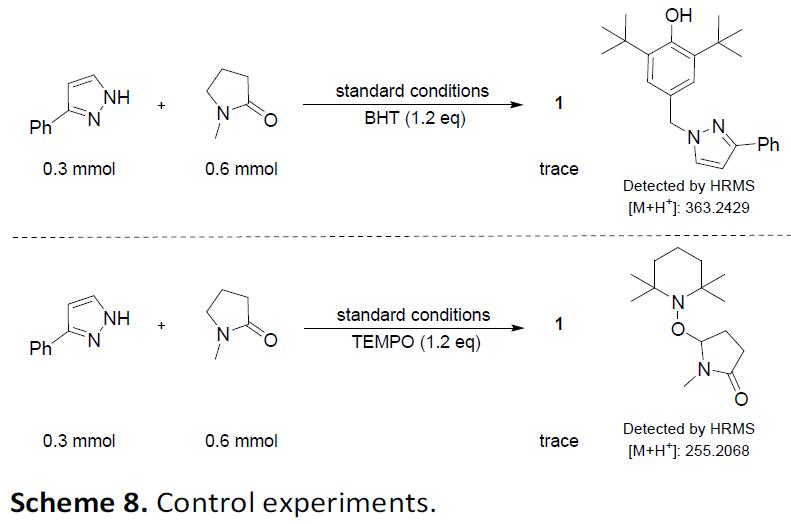
为了进一步确定反应的机理,作者进行了一些对照实验(Scheme 8)。将2,2,6,6-四甲基哌啶-1-氧基(TEMPO)和2,6-二叔丁基苯酚(BHT)分别加入电化学氧化反应中,可抑制反应的发生。同时,HRMS检测到两种产物。从而表明,该反应可能涉及自由基参与。
为了进一步详细了解机理,作者进行了循环伏安法(CV)实验(Scheme 9)。反应结果表明,3-苯基-1H-吡唑的氧化峰(2.04 V,红线)小于NMP的氧化电位(2.39 V,蓝线)。因此,3-苯基-1H-吡唑在反应中首先被氧化。随后,作者又进行了电子顺磁共振(EPR)实验,以确定自由基中间体。当体系存在1-甲基-2-吡咯烷酮时,未检测到自由基信号。这些结果证明,3-苯基-1H-吡唑作为被阳极氧化的第一个底物。

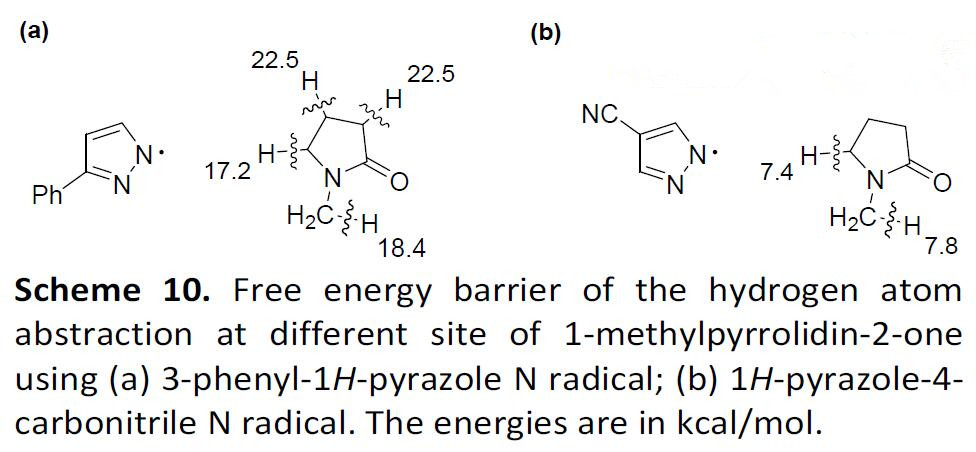
此外,作者通过DFT计算,使反应的选择性更加合理化(Scheme 10)。当使用3-苯基-1H-吡唑作为底物时,抽氢过程中的最低自由能障发生在C3-H和N-CH2-H的位上,与实验观察到的选择性非常吻合。同样使用1H-吡唑-4-腈作为底物时,选择性的趋势也相同。
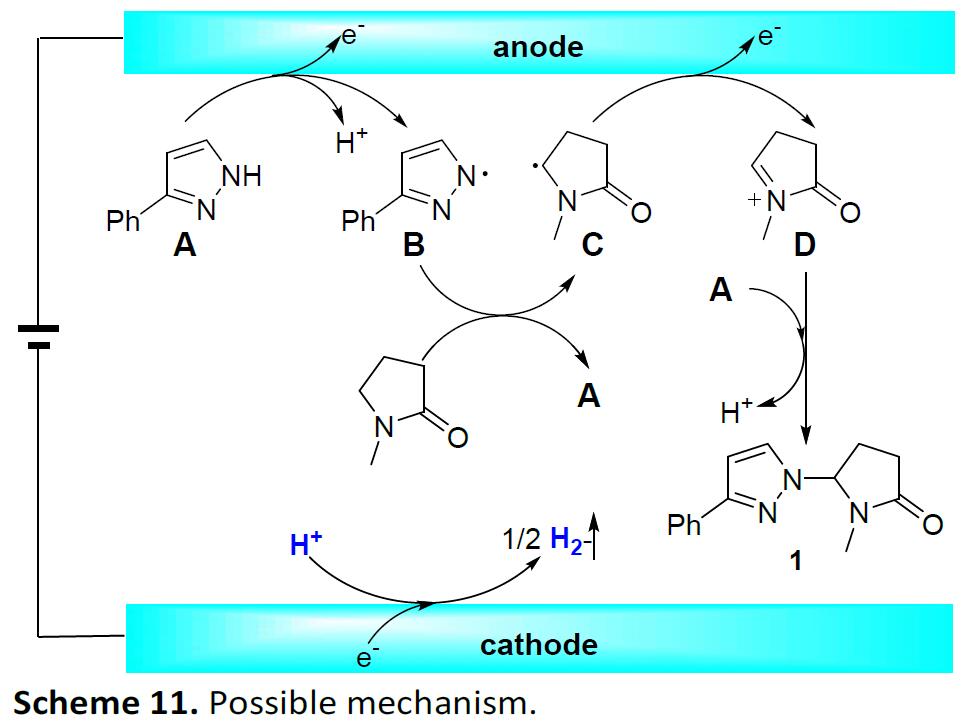
根据上述实验以及文献的查阅,作者提出了一种可能的反应机理(Scheme 11)。首先,具有较低氧化电位的底物A在阳极被氧化,然后进行氢转移形成氮自由基B。紧接着,自由基B和1-甲基-2-吡咯烷酮经历分子间氢转移,形成自由基C和底物A。随后,自由基C在阳极上被氧化,生成亚胺阳离子D。最后,D再与底物A反应,获得产物1。同时,质子氢在阴极还原时产生氢气。
总结
武汉大学雷爱文教授课题组报道了在无金属催化剂和外部氧化剂的条件下,通过电化学氧化,实现吡咯与内酰胺化合物的直接C(sp3)-H杂环化反应。该反应具有广泛底物范围、温和的反应条件等优点。克级实验以及药物的后期修饰,进一步证明了反应的实用性。此外,对照实验、循环伏安实验、DFT计算和EPR实验,进一步证明了机理的正确性。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!















No comments yet.