

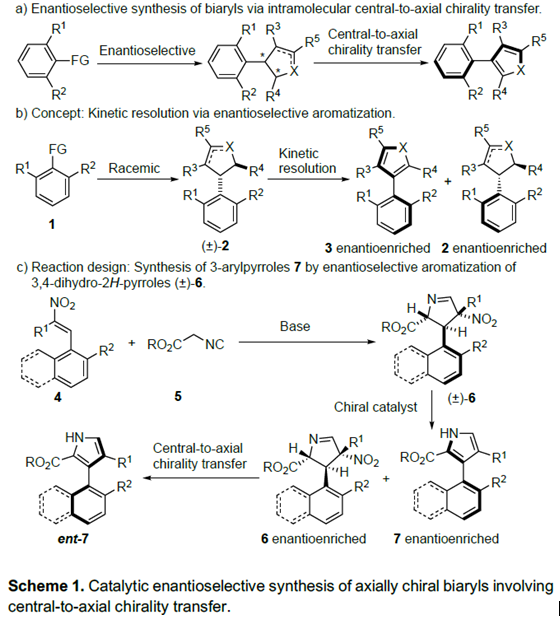
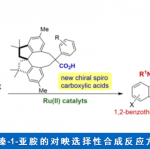
轴手性化合物广泛存在于天然产物、药物、配体及催化剂中,因此合成手性联芳基化合物一直是化学家关注的重中之重。合成轴手性化合物的方法主要有4种:a)两个芳基单位发生不对称偶联反应; b)已存在的联芳基化合物去对称化;c)取代芳基化合物的阻转选择性; d)联芳基化合物的动力学拆分。Scheme 1a则是此前报道的取代芳基化合物的不对称加成产物发生手性中心向轴手性的传递过程,得到含五元杂环的轴手性化合物。这种合成轴手性的方法很早就被提出,且取得了不错的进展,但关于通过自身转化构建含五元杂环的轴手性骨架的报道还比较少。另一方面,外消旋二芳基化合物的动力学拆分可通过醇解、N-酰化/烷基化、O-酰化以及直接芳基官能团化实现,但到目前为止,还没有关于外消旋非二芳基化合物的动力学拆分的报道。最近,瑞士洛桑联邦理工大学祝介平课题组报道了异氰乙酸酯和炔基酮的不对称杂环反应,构建轴手性芳基吡咯骨架[1]。在此基础上,作者想要将动力学拆分引入轴手性芳基吡咯化合物的合成中,即官能团取代的芳环发生环化反应可得到消旋的产物2。在合适的催化剂作用下,2可发生氧化反应或消除反应从而实现2的动力学拆分过程,得到轴手性化合物3。同时,回收的2可转化为反构型的产物3(Scheme 1b)。3,4-二氢-2H-吡咯6为Barton-Zard反应的中间体,作者推测6可成功实现动力学拆分过程。因此,祝介平课题组成功开发了碱性催化作用下硝基烯烃4与α-异氰基乙酸酯5发生环化反应生成消旋产物6。在奎宁衍生的硫脲催化剂作用下,作者成功实现了6的动力学拆分,合成轴手性芳基化合物(Scheme 1c)。
Catalytic Kinetic Resolution via Enantioselective Aromatization: Conversion of Racemic Intermediates of the Barton-Zard Reaction to Enantioenriched 3-Arylpyrroles
Zheng, S.-C.; Wang, Q.; Zhu, J.* Angew. Chem. Int. Ed. 2019, Just Accepted Manuscript
DOI: 10.1002/anie.201903589
论文作者介绍:

论文作者:祝介平教授
教育背景:
- 1984, B.Sc., Hanzhou Normal University, P. R. China
- 1987, M.Sc., Lanzhou University (Prof. Y.-L. Li), P. R. China
- 1991, Ph.D., Université Paris XI (Prof. H.-P. Husson and Prof. J.-C. Quirion), France
- 1991-1992, Post-doct., Texas A & M University (Prof. Sir D. H. R. Barton), USA Academic Positions
- 1992-2000, “Charge de Recherche” at ICSN, CNRS, France
- 2000-2006, Director of Research, 2nd class, at ICSN CNRS, France
- 2006-2010, Director of Research, 1st class at ICSN, CNRS, France 2010-September 1st- Professor, ISIC, EPFL
获奖情况:
- 1996, CNRS bronze medal (1996)
- 1999, French Chemical Society SCF-Acros award (1999)
- 2002, AstraZeneca Award in Organic Chemistry (UK)
- 2002, Japan Society for Promotion of Science (JSPS) fellow
- 2003, Prix Emile Jungfleisch of French Academy of Sciences
- 2003, National Science Foundation Outstanding Young Oversea Scientist award (China)
- 2004, Liebig Lectureship of the German Chemical Society
- 2008, Novartis Chemistry Lecture Award (Switzerland)
- 2009, CNRS Silver medal
- 2010, French Chemical Society-Division of Organic Chemistry SCF-DCO award. 2015, Nankai University Lectureship on Organic Chemistry, Nankai University, Tianjing, P. R: China
- 2016, Royal Society of Chemistry (RSC) Natural Product Chemistry Award, UK
- 2017, KAIST-BK21 School of Molecular Science Lectureship award, South Korea
研究领域:
主要从事复杂天然产物全合成、不对称催化与合成和多组分串联反应领域的研究。
论文概要:
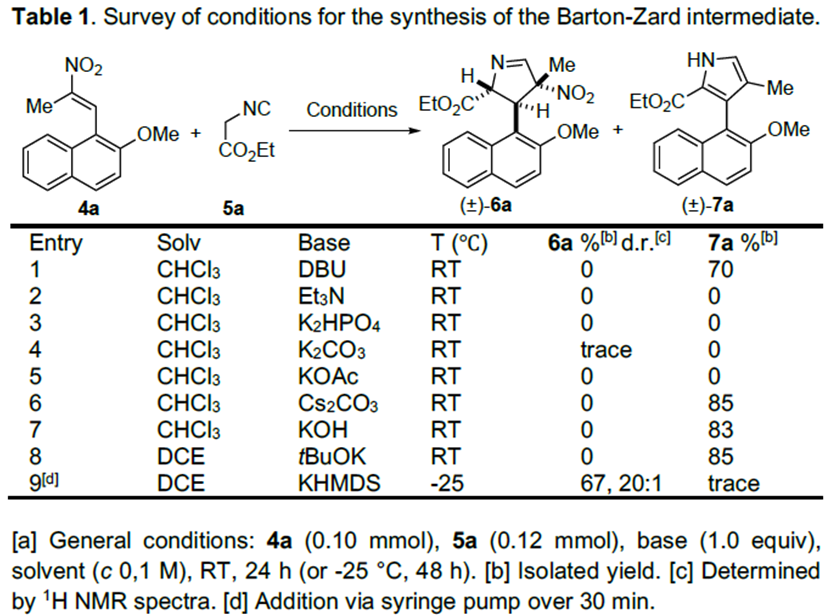
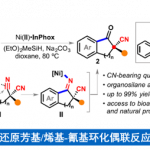
文献调研显示已有关于硝基烯烃4与α-异氰基乙酸酯5发生[3+2]环加成反应合成2,3-二氢吡咯的报道,但由于在DBU或胍催化作用下,产物会迅速脱掉HNO2,所以未分离出中间体6。因此,为分离出中间体6a,作者以4a和5a为模板底物,对反应条件进行反复筛选,确定最佳条件为(Table 1):1.0 equiv KHMDS(0.5 N在甲苯中)为最优碱,DCE为最优溶剂,在-25 °C条件下反应48 h,能以67%的收率得到中间体6a。
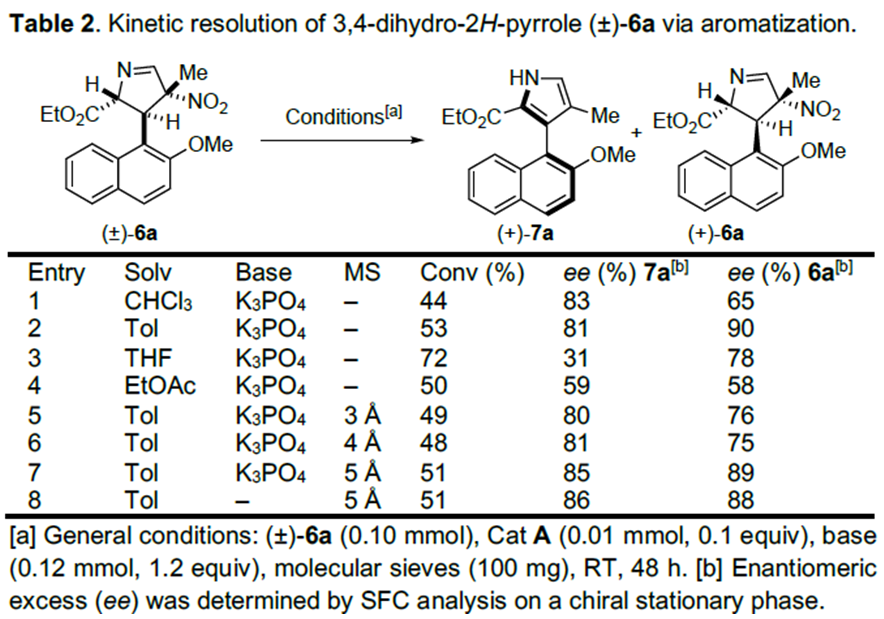
紧接着,作者对(±)-6a的动力学拆分条件进行优化,发现0.1 equiv奎宁衍生物硫脲A为催化剂,1.2 equiv K3PO4为碱,100 mg 5Å分子筛为添加物,在室温条件下反应48 h,能以51%的收率和86%的对映选择性得到相应产物(+)-7a;同时,可以49%的收率和88%的对映选择性回收(+)-6a(Table 2) 。
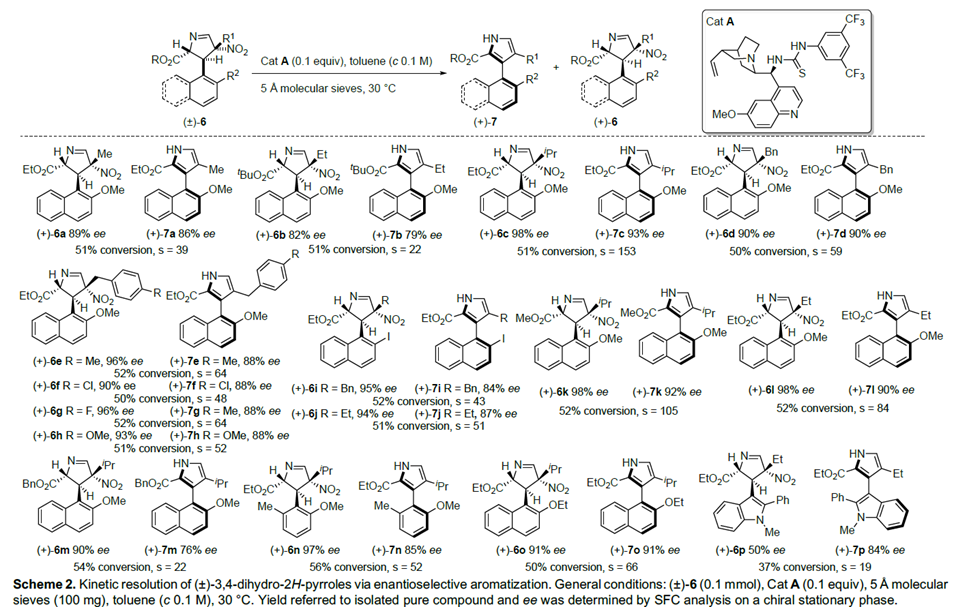
在最优反应条件下,作者对该动力学拆分的底物范围进行考察(Scheme 2)。各种萘基取代、C-4位甲基、乙基、异丙基、苄基取代、C-2位甲氧基羰基、乙氧基羰基取代吡咯、3-(2′-甲氧基-6′-甲基苯基)吡咯均能较好的适应反应条件,能以较高的收率和对映选择性得到相应产物。而C-2位叔丁氧基羰基和苄氧基羰基取代的吡咯的对映选择性略微有所降低。带两个5元杂环的吡咯以及(±)-3,4-二氢-2H-吡咯也能适应反应条件,但其收率较低。
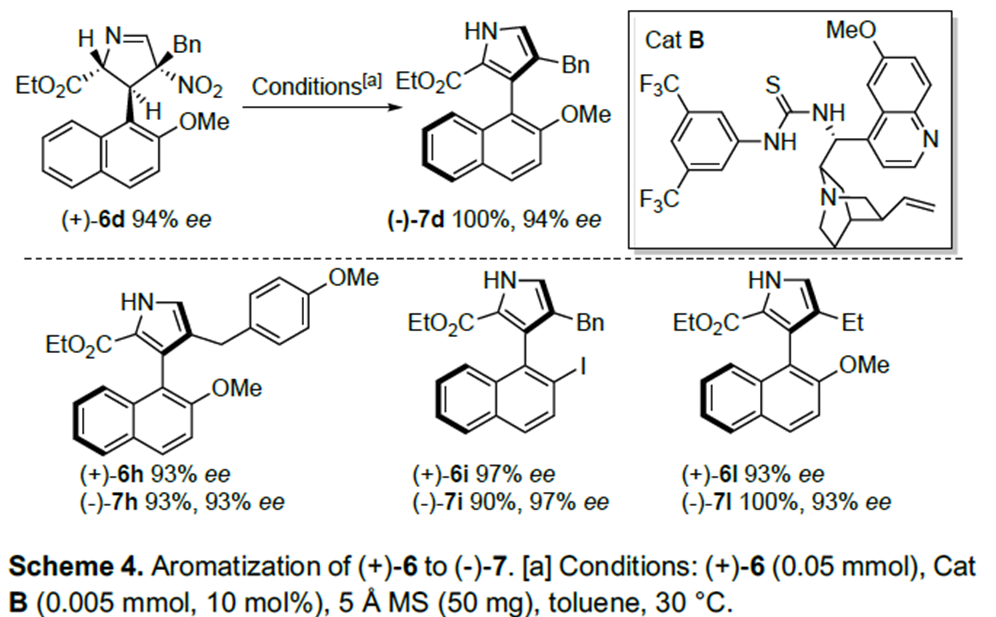
以奎尼丁衍生的硫脲B为催化剂,5Å分子筛为添加物,甲苯为溶剂, 30°C条件下,(+)-6d能以较高的对映选择性转化为(-)-7d(Scheme 4)。
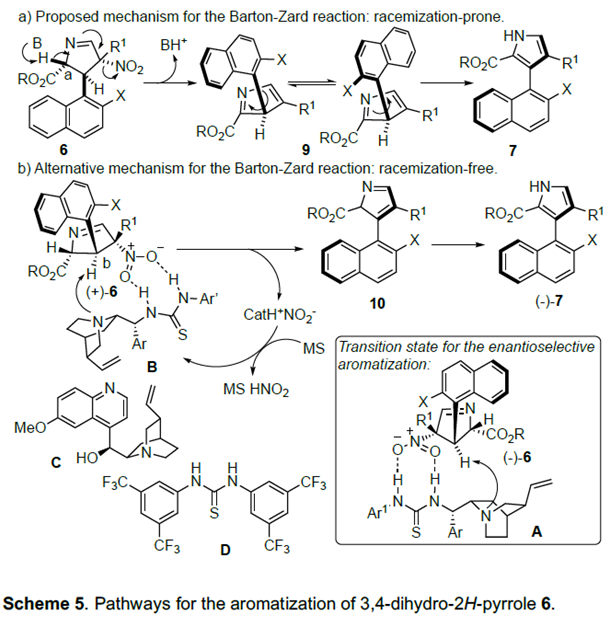
大家所熟知的联芳基化合物的动力学拆分机理为:碱性作用下生成的中间体6的酸性Ca-H会发生去质子化作用,然后发生NO2消除反应得到3H-吡咯9。9再发生芳构化反应会转化为7。但当DBU或Cs2CO3为碱时,9的Csp3-Csp2键容易旋转,导致产物部分消旋化(Scheme 5a)。在此基础上,作者通过实验观察推测该反应可能的机理:催化剂的硫脲基团可与硝基形成H-键,从而使B的叔胺基离Cb-H足够近。在催化剂B的作用下,催化剂-6复合体发生顺式消除生成2H-吡咯10,紧接着发生芳构化反应转化为7。空间位阻作用会阻碍10的Csp2-Csp2键旋转,因此,该反应的外消旋化可降到最低(Scheme 5b)。
论文总结评价:
瑞士洛桑联邦理工大学祝介平课题组报道了第一例硝基烯烃与α-异氰基乙酸酯发生Barton-Zard反应,得到的消旋2-芳基吡咯在金鸡纳生物碱衍生双功能催化剂作用下可顺利的发生动力学拆分过程。这是第一例外消旋化合物通过不对称芳构化反应实现动力学拆分,合成手性联芳基化合物的报道。
参考文献:
- Zheng, S.-C.; Wang, Q.; Zhu, J. Chem. Int. Ed. 2019, 58, 1494. DOI: 10.1002/anie.201812654
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!













No comments yet.