

这次介绍的论文报道了简短路线的Paspaline A的全合成、以及Emindole PB的首次全合成。通过使用DFT计算,高效确定了中间体的最佳结构,减少了不必要的尝试,节约了时间。
用计算化学评价反应的可行性及其在天然产物合成中的应用
逆合成分析法是在化合物的合成路线设计中最常用的一种理论方法。但是,对于简化合成的逆合成分析断键法来说,由于可参考的文献是不充足的,所以很多还需要通过实际的实验验证其可行性。现代的电脑计算,逐渐转变成这种逆合成分析的强力的手段之一。换句话说,通过算法的开发可以有效地识别化学键断裂与通过量子化学计算实现前所未有的合成路线的设计成为可能。
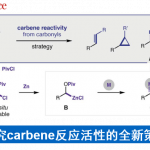
另一方面,吲哚二萜类化合物一直是作为全合成研究目标的天然产物群。Paspaline A(1)具有六环骨架,且含有与连续季碳相邻的吲哚稠合环戊烷结构。该类天然产物具有抗哺乳动物乳腺癌的抗增殖・抗转移活性。在迄今已报道的1的全合成中,采用了吲哚环与D环之间的环戊酮部分(C环)的构建的合成策略、合成步骤多(图1A)[2,3]。另外、Emindole PB (2)的全合成还未有报道。
这次、耶鲁大学的Newhouse副教授课题组、模仿生物合成的合成策略(图1C)、通过使用吲哚的亲核性来构建C环,实现了短程全合成。此外,通过对相同的中间体进行甲基重排,成功实现了首次Emindole PB (2)的全合成和结构测定(图1B)。通过使用密度泛函理论(DFT)计算确定C环的环化前体的最佳结构,成功地降低了最优化合成路线的筛选上的耗时。
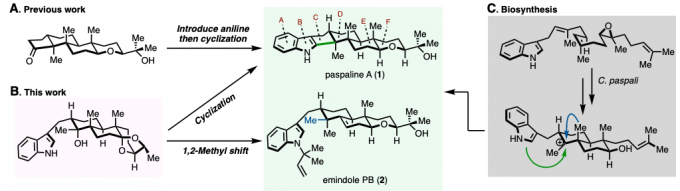
图1. (A) Paspaline A的全合成报道 (B) 这次的合成策略 (C) 生物合成
“Total Synthesis of Paspaline A and Emindole PB Enabled by Computational Augmentation of a Transform-Guided Retrosynthetic Strategy”
Kim, D. E.; Zweig, J. E.; Newhouse, T. R. J. Am. Chem. Soc.2019, ASAP. DOI: 10.1021/jacs.8b13127
论文作者介绍

履历:
2001-2005 B.A., Colby College, ME, USA (Prof. Dasan M. Thamattoor)
2006-2010 Ph.D, The Scripps Research Institute, CA, USA (Prof. Phil S. Baran and Prof. Donna G. Blackmond)
2010-2013 Posdoc,Harvard University, MA, USA (Prof. E. J. Corey)
2013-2018 Assistant Prof. at Yale University, CT, USA
2018- Associate Prof. at Yale University
研究内容:计算化学辅助天然产物全合成、过渡金属催化的反应方法学开发及全合成
论文概要
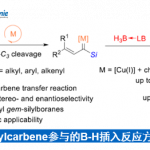
Newhouse副教授等人、首先将吲哚与二萜骨架连接,并计划使用吲哚的亲核性通过C环构建来实现1的合成。通过对位于远端位的具有不同F环前体结构的三种化合物的三级碳正离子中间体Z(a)–Z(c)进行DFT计算来比较它们的反应选择性,来预测最容易环化的底物结构(图2A)。具体而言,是计算了三级碳正离子中间体环化与甲基重排反应进行时的能量壁垒的差。结果发现,当使用在F环上具有双环缩酮结构Z(c)时,甲基重排与环化的能垒之间的差最大(-4.5kcal / mol),因此作者决定通过经由该中间体合成1。
作者从Wieland-Miescher 酮衍生物出发,5步合成了环化前体3、然后在AlCl3作用下、得到了C环环化体4与甲基重排体5(4:5= 1:3)(图2B)。4在还原条件下诱导合成了Paspaline A(1)(合计9步反应)。另一方面、从5开始,分4步进行烯烃的异构化和F环的形成,最后,将氮原子叔丁基化,得到Emindole PB (2)(共14步)。
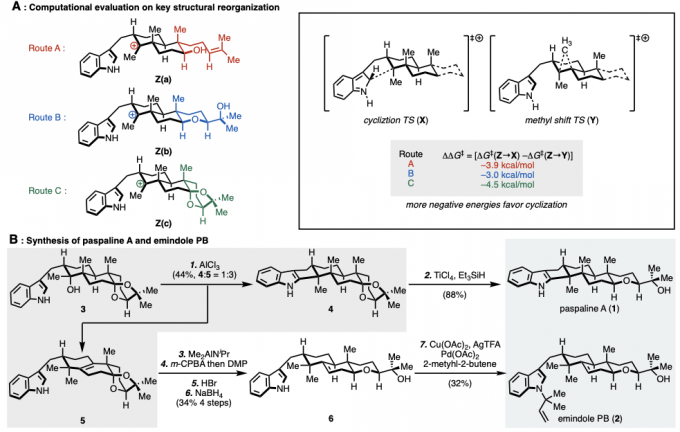
图2. (A)DFT计算 (B)Paspaline A以及Emindole PB的合成
综上、本次作者以1/3的步骤数合成了Paspaline A与Emindole PB。通过DFT计算确定了最优底物,节省了时间。尽管这种计算方法的能力和局限性是未知的但非常期待通过未来研究的进一步发展,使得这种方法变得更可靠。
参考文献
- Corey, E. J.; Wipke, W. T. Science 1969, 166, 178–192.DOI: 1126/science.166.3902.178
- (a) Smith, A. B.; Mewshaw, R. J. Am. Chem. Soc. 1985, 107, 1769–1771. DOI: 10.1021/ja00292a058 (b) Smith, A. B.; Leenay, T. L. J. Am. Chem. Soc. 1989, 111, 5761–5768. DOI: 10.1021/ja00197a039
- Sharpe, R. J.; Johnson, J. S. J. Am. Chem. Soc. 2015, 137, 4968–4971. DOI: 10.1021/jacs.5b02631
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!



















No comments yet.