

本文作者:杉杉
导读
有机磷化合物已经广泛应用于化学、材料科学以及生物学领域的相关研究。因此,设计更为高效的构建C-P键的全新策略,在有机合成化学研究中具有重要意义。其中,前期已经报道的一系列在非电化学或电化学条件下进行的C-H膦酸酯化反应 (C-H phosphorylation)方法学的相关研究中,通常需要引入相应的导向基团、过渡金属催化剂或化学氧化剂,同时,底物应用范围较为有限。为解决上述问题,近日,厦门大学的徐海超课题组报道一种在无催化剂与无外部氧化剂以及连续流动的条件下进行的电化学芳香C-H膦C-H化反应方法学,进而成功实现一系列芳基膦C-H类化合物的构建。在这一全新的电化学芳香C-H膦酸酯化策略中,通过芳基化合物与阳极产生的P-自由基正离子之间的反应过程,进而形成相应的C-P键。同时,通过具有高度反应活性的P-自由基正离子与温和的有机电合成反应条件的结合,进而使一系列具有不同电子特性的芳基化合物能够有效地参与上述的膦酸酯化过程。之后,该小组这一全新的膦酸酯化策略进一步应用于各类复杂天然产物与生物活性化合物的选择性后期官能团化过程的相关研究。此外,作者进一步发现,这一全新的电化学芳香C-H膦酸酯化策略同样能够顺利完成相应的克级规模反应,进而表明这一策略具有潜在的合成应用价值。
Electrochemical C-H phosphorylation of arenes in continuous flow suitable for late-stage functionalization
H. Long, C. Huang, Y. Zheng, Z. Li, L. Jie, J. Song, S. Zhu, H. C. Xu, Nat. Commun. 2021, ASAP doi: 10.1038/s41467-021-26960-y.
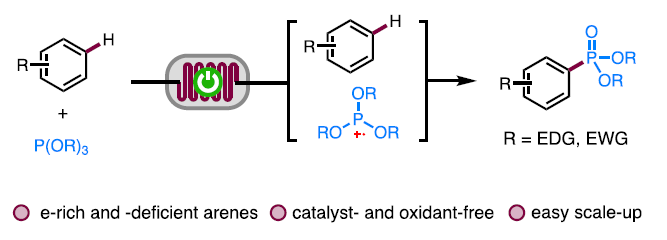
正文
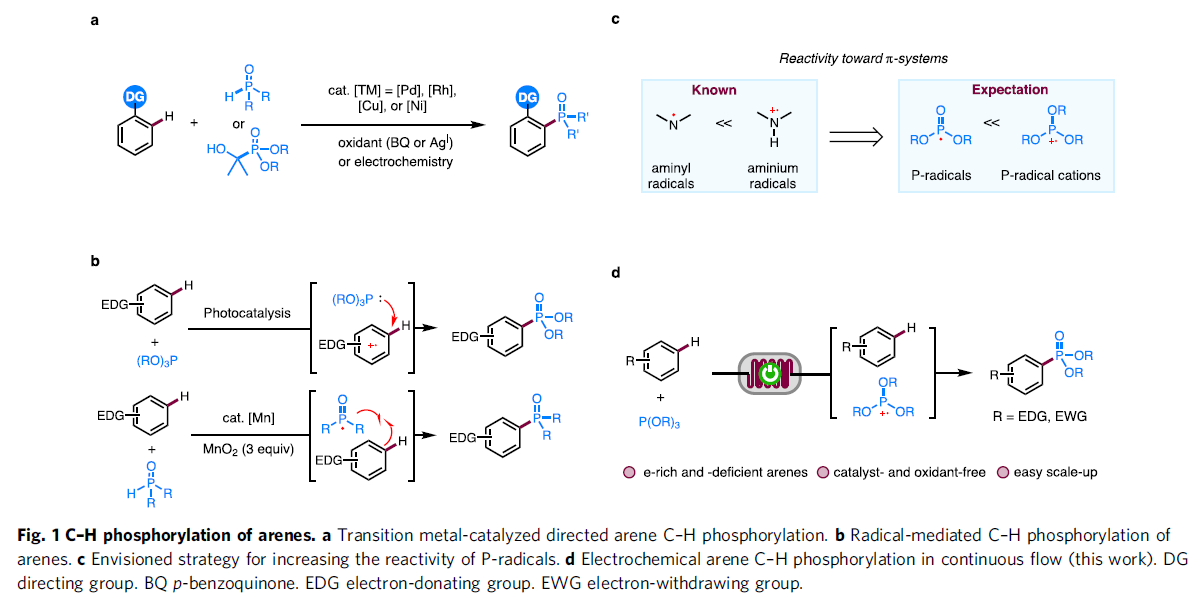
芳基磷化合物目前已经广泛应用于医药化学、材料科学以及催化配体与Lewis酸催化领域的相关研究。与传统的Hirao反应方法学[1]相比,C-H膦酸酯化策略更加具有吸引力,并且能够有效地避免相关底物的预官能团化步骤。目前,对于非电化学条件下的芳香C-H膦酸酯化方法学的相关研究 (Fig. 1a-b),已经有诸多的文献报道[2]-[4]。然而,上述的合成转化过程中,通常需要加入过量的芳基底物,而且底物应用范围十分有限。
同时,近年来,电化学条件下的各类富电子杂芳基化合物[5]以及具有导向基团的芳基化合物[6]的C-H膦酸酯化方法学的相关研究已经取得较大进展。然而,上述的电化学C-H膦酸酯化策略对于缺电子的芳基底物,则需要采用各类金属催化剂,同时需要选择具有三电极配置的无隔膜电解槽[7]-[11]。
接下来,受到通过胺基自由基 (aminyl radical)质子化形成的胺正离子自由基 (aminium radical)能够显著提高π-体系反应活性的相关研究报道[12]-[13]的启发,作者设想,与P-自由基相比,P-自由基正离子具有更高的反应活性,进而能够与各类缺电子芳基底物进行有效的反应 (Fig. 1c)。同时,作者进一步设想,采用连续流动的电化学反应器,可能更加有利于P-自由基正离子的形成,并通过芳基底物的捕获,形成相应的芳香C-H膦酸酯化产物。基于上述的研究设想,厦门大学的徐海超团队成功设计出一种在连续流动条件与无催化剂以及无外部氧化剂存在条件下进行的电化学芳香C-H膦酸酯化的反应方法学,进而顺利完成一系列芳基膦酸酯分子的构建 (Fig. 1d)。
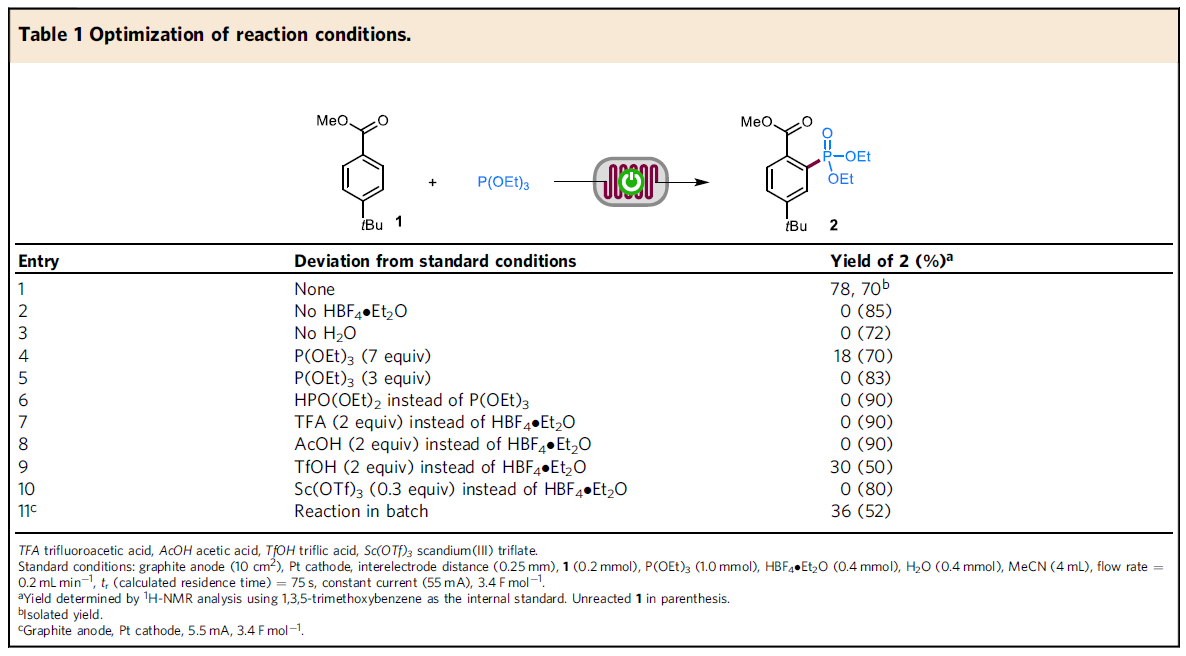
首先,作者采用1与P(OEt)3作为模型底物,进行相关膦酸酯化反应条件的优化筛选 (Table 1)。进而确定最佳的反应条件为:在石墨作为阳极,Pt作为阴极的无隔膜连续流动电解槽中,采用HBF4作为酸性添加剂,H2O作为添加剂,MeCN作为反应溶剂,流速为0.2 ml/min-1,控制电流为45-55 mA,最终获得70%收率的膦酸酯化产物2。
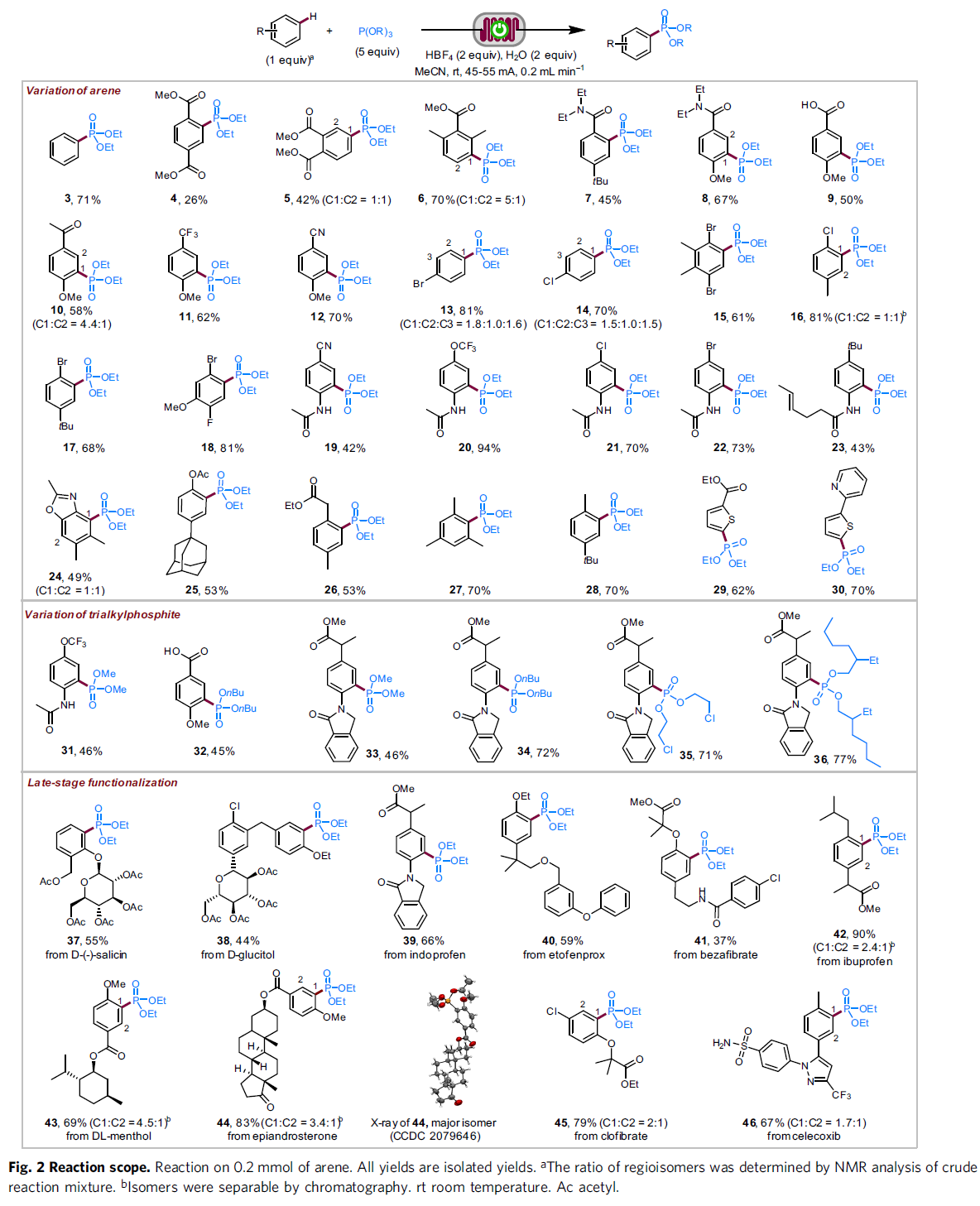
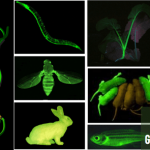
在上述的最佳反应条件下,作者首先对一系列芳基底物的应用范围进行考察 (Fig. 2)。研究表明,一系列带有供电子与吸电子基团取代的芳基底物均能较好地与上述的标准反应条件兼容,并获得相应的膦酸酯化产物3–28 (26-94% 收率)。值得注意的是,C-P键形成过程的区域选择性与Friedel-Crafts反应类似,即芳基底物中无高度立体位阻基团存在时,反应优先在供电子基团的邻位或对位进行。之后,作者发现,2-取代噻吩底物,同样能够有效地完成上述的膦酸酯化过程,并获得相应的4-膦酸酯产物29与30。接下来,该小组进一步对一系列亚磷酸酯的底物应用范围进行深入研究。作者发现,一系列具有不同一级烷基链的亚磷酸三烷基酯底物,均能够顺利地参与上述的膦酸酯化过程,并获得相应的目标产物31–36 (45-77% 收率)。此外,研究发现,这一全新的膦酸酯化策略同样能够良好地应用于各类复杂天然产物以及生物活性化合物 (37–46)的后期官能团化过程。同时,该小组进一步观察到,上述的标准反应条件对于一系列具有多重芳环存在的芳基化合物,其膦酸酯化过程则优先选择在最为富电子的芳环中进行 (38、40、41与46)。
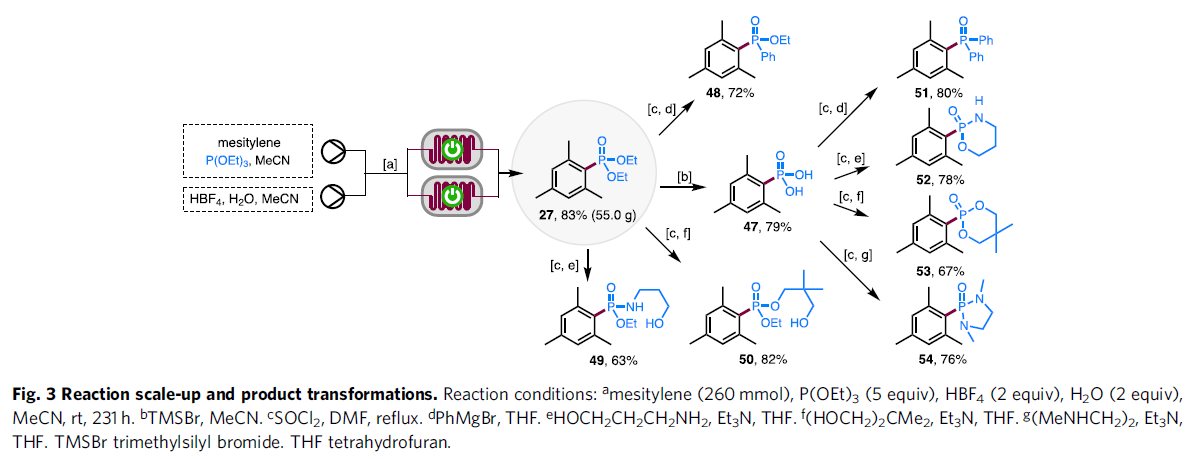
之后,作者进一步对上述膦酸酯化策略的合成实用性进行研究 (Fig. 3)。首先,该小组发现,将均三甲苯底物、P(OEt)3的MeCN溶液以及HBF4添加剂、H2O-MeCN混合溶剂分别通过采用两重平行的连续流动电解槽构成的反应器进行充分混合之后,能够顺利完成克级规模 (214 mmol的)膦酸酯化反应,并以良好的反应收率 (83%),获得相应的三甲苯基膦酸酯产物 27。同时,作者进一步观察到,通过上述膦酸酯化策略获得的甲基膦酸酯产物27能够有效地转化为其它不同类型的有机磷砌块47-54。
接下来,为提出合理的反应机理,作者进行一系列相关的实验研究 (Fig. 4)。首先,作者通过P(OEt)3、芳基底物1以及HPO(OEt)2的CV实验表明,在上述的三种有机磷分子中,P(OEt)3 (Ep/2 = 1.50 V vs SCE)能够最为容易地参与相应的氧化过程 (Fig. 4a)。同时,作者通过对芳基底物1与P(OEt)3的反应混合物,在上述的标准反应条件以及无水存在时的相关31P-NMR谱图分析 (Fig. 4b)表明,P(OEt)3在无水存在的条件下,能够分解为多种不同类型的有机含磷中间体。相反地,在标准条件下,通过31P-NMR分析,则能够成功指认出三种主要的有机磷中间体,即化合物2、HPO(OEt)2与OP(OEt)3。同时,由于亚磷酸三烷基酯在酸的促进下,能够迅速水解,形成相应的H-磷酸酯 (H-phosphonate),因此,作者观察到,在P(OEt)3与HPO(OEt)2存在以及未加入水的条件 (反应条件II与 III)下,采用芳基底物1进行的膦酸酯化过程中,在2 eq. HPO(OEt)2存在下,能够获得58%的反应收率;而在0.2 eq. HPO(OEt)2存在下,则能够获得更加优良的反应收率 (78%, Fig. 4b)。同时,该小组发现,在条件II与 III中,反应混合物的31P-NMR谱图与标准反应条件下的31P-NMR谱图十分类似。
之后,作者进一步发现,芳基底物1在P(OEt)3与HPO(OnBu)2存在下进行的电解过程中,则获得51%收率的膦酸酯化产物2以及痕量的产物55 (Fig. 4c),进而表明膦酸酯产物中的PO(OR)2基团源自于P(OR)3而非HPO(OR)2。同时,该小组发现,反应混合物中加入H218O时,在膦酸酯化产物2中并未检测出18O 的存在 (Fig. 4d)。综上实验观察表明,HPO(OR)2的存在对于C-H膦酸酯化反应的顺利进行尤为关键。然而,反应过程中HPO(OR)2的具体作用则仍有待深入研究。此外,作者进一步通过苯与苯-d6之间分子间竞争反应实验,观察到KIE为1.0,进而表明,电解反应的决速步骤中并未涉及C(aryl)-H键的断裂 (Fig. 4e)。

基于上述研究,作者提出一种可能的反应机理 (Fig. 4f)。首先,通过亚磷酸三烷基酯56的阳极氧化过程,形成相应的P-自由基正离子57,再通过57与芳基底物58作用,形成相应的远程自由基正离子59。之后,59经历进一步的阳极氧化以及后续的去质子化过程,形成phosphonium中间体60。接下来,通过P(OR)3水解或反应后处理过程中产生的水或醇分子对于中间体60的亲核进攻过程,获得相应的膦酸酯化产物61。同时,在酸性条件下,反应混合物中大量存在的质子能够在Pt阴极进行进一步的还原析氢过程,形成H2。此外,酸性添加剂HBF4除能够促进P(OR)3的水解之外,同样能够作为支持电解质以及析氢过程中的质子源,进而有效避免缺电子的中间体60以及目标分子61进一步参与后续的阴极还原过程。并且,通过P(OR)3水解原位形成的HPO(OR)2中间体,同样可能与自由基正离子57通过后续的可逆反应步骤,形成相应的加合物62,进而有效地减弱57的进一步分解。因此,自由基正离子57能够顺利地参与后续的芳香C-H膦酸酯化过程。
总结
厦门大学的徐海超教授课题组在连续流动的反应条件下,成功设计出一种全新的芳香C-H膦酸酯化反应方法学。反应过程中,无需加入相应的分子催化剂以及传统的化学氧化剂。同时,这一全新的C-H膦酸酯化策略具有反应条件温和、底物应用范围广泛、高度的官能团兼容性高等优势。
参考文献
[1] C. S. Demmer, N. Krogsgaard-Larsen, L. Bunch, Chem. Rev. 2011, 111, 7981. doi: 10.1021/cr2002646. [2] (a) C. G. Feng, M. Ye, K. J. Xiao, S. Li, J. Q. Yu, J. Am. Chem. Soc. 2013, 135, 9322. doi: 10.1021/ja404526x.(b) C. Li, T. Yano, N. Ishida, M. Murakami, Angew. Chem. Int. Ed. 2013, 52, 9801. doi: 10.1002/ange.201305202.
[3] L. Niu, J. Liu, H. Yi, S. Wang, X. Liang, A. Singh, C. Chiang, A. Lei, ACS Catal. 2017, 7, 7412. doi: 10.1021/acscatal.7b02418. [4] O. Berger, J. L. Montchamp, J. Org. Chem. 2019, 84, 9239. doi: 10.1021/acs.joc.9b01239. [5] S. Wang, Q. Xue, Z. Guan, Y. Ye, A. Lei, ACS Catal. 2021, 11, 4295. doi: 10.1021/acscatal.1c00549. [6] Z. Wu, F. Su, W. Lin, J. Song, T. Wen, H. Zhang, H. Xu, Angew. Chem. Int. Ed. 2019, 58, 16770. doi: 10.1002/anie.201909951. [7] S. O. Strekalova, V. V. Grinenko, T. V. Gryaznova, A. I. Kononov, E. L. Dolengovski, Y. H. Budnikova, Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 506. doi: 10.1080/10426507.2018.1540488. [8] M. N. Khrizanforov, S. O. Strekalova, K.V. Kholin, V. V. Khrizanforova, M. K. Kadirov, T. V. Gryaznova, Y. H. Budnikova, Catal. Today 2017, 279, 133. doi: 10.1016/j.cattod.2016.06.001. [9] E. O. Yurko, T. V. Gryaznova, Y. H. Budnikova, Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 343. doi: 10.1080/10426507.2018.1541897. [10] M. N. Khrizanforov, S. O. Strekalova, V. V. Grinenko, V. V. Khrizanforova, T. V. Gryaznova, Y. H. Budnikova, Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1491. doi: 10.1080/10426507.2016.1212051. [11] Y. H. Budnikova, Y. B. Dudkina, Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 415. doi: 10.1080/10426507.2018.1540480. [12] A. Ruffoni, J. Fabio, T. D. Svejstrup, A. J. McMillan, J. J. Douglas, D. Leonori, Nat. Chem. 2019, 11, 426. doi: 10.1038/s41557-019-0254-5. [13] J. M. Ganley, P. R. D. Murray, R. R. Knowles, ACS Catal. 2020,10, 11712. doi: 10.1021/acscatal.0c03567.


















No comments yet.