
本文作者:杉杉
导读
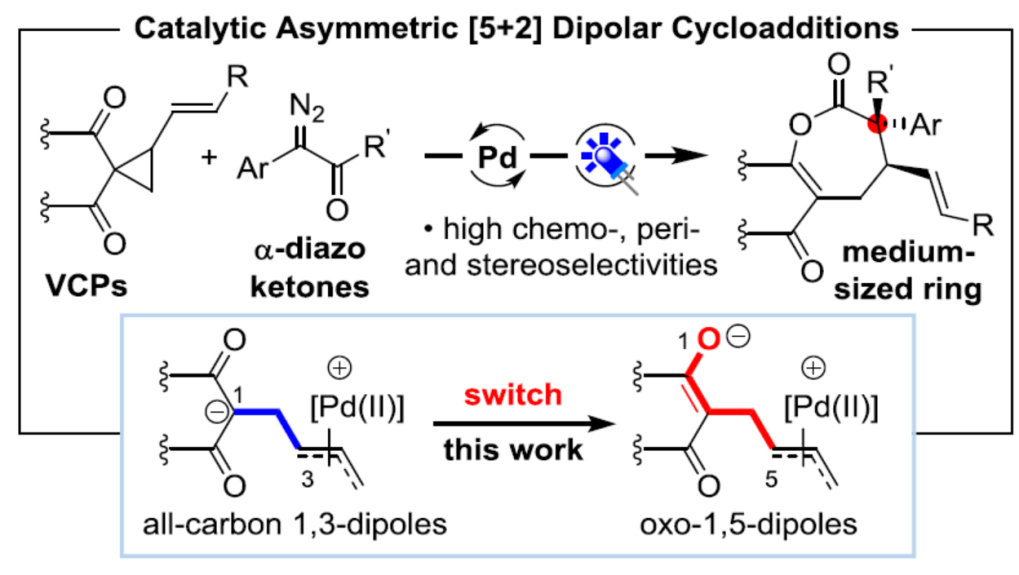
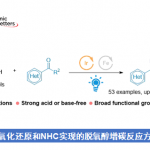
乙烯基环丙烷(VCPs)衍生物广泛应用于过渡金属催化的环加成反应中,由于可快速构建各类新型环状骨架,因此在现代有机合成化学中具有重要的意义。近日,华中师范大学陆良秋教授课题组和重庆大学蓝宇教授(理论计算)合作共同在Angew. Chem. Int. Ed.上发表论文,通过钯催化(可见光驱动),首次实现VCPs与α-重氮酮的不对称[5+2]环加成反应,获得23个七元环内酯化合物(收率高达92%,er高达99:1,dr高达12.5:1)。同时,该反应中涉及含钯偶极中间体由全碳1,3-偶极子向oxo-1,5-偶极子的转化的过程,从而使反应活性发生改变。此外,通过相关的计算化学研究同样能够对所观察到的周边选择性(periselectivity)进行合理解释。
Exploitation of the New Reactivity of Vinylcyclopropanes for Palladium-Catalyzed, Asymmetric [5+2] Dipolar Cycloadditions
M.-M. Li, Q Xiong, B.-L. Qu, Y.-Q. Xiao, Y. Lan*, L.-Q. Lu,*and W.-J. Xiao
Angew. Chem. Int. Ed. 2020, 59, 17429 –17434 DOI: 10.1002/anie.202006366. https://doi.org/10.1002/anie.202006366
正文:
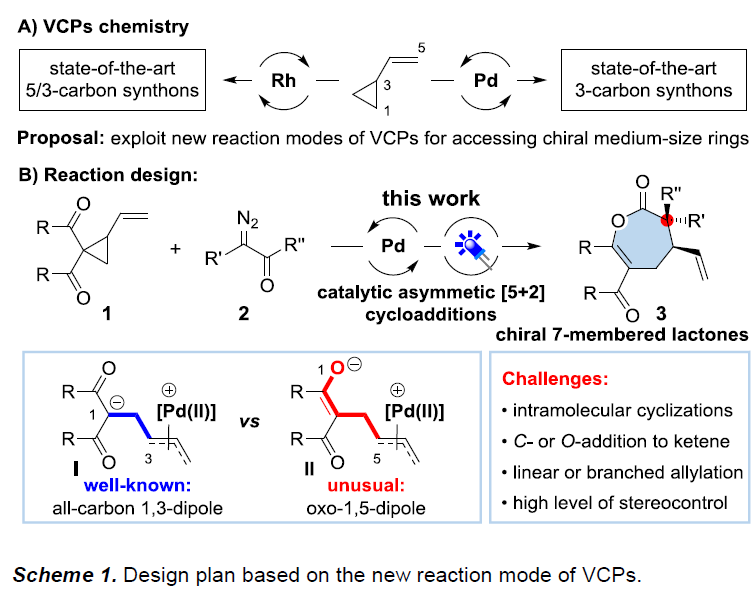
近几十年来,过渡金属催化乙烯基环丙烷(VCPs,vinylcyclopropanes)的环加成反应已经有广泛研究,进而获得各类环状化合物。在Rh催化环加成反应中,VCP可以作为5碳合成子[1](如[5+1],[5+2]和[5+2+1]环加成)或3碳合成子[2](如[3+2]和[3+2+1]环加成)(Scheme 1A, left)。由于环丙烷环中缺乏活化基团,因此,多数VCP可视为电中性的合成子。1985年,Tsuji课题组发现[3],偕双吸电子基活化的VCP可在Pd催化下与缺电子的烯烃发生[3+2]环加反应。反应过程中,Pd(0)催化剂通过形成具有烯丙基-Pd配位的偶极子,再与VCP进一步反应,进而完成[3+2]环加成过程。在之后的几十年中,几乎所有Pd催化VCP活化的文献均获得[3+2]环加成产物(Scheme 1A, right)。在此,华中师范大学陆良秋教授课题组将活化的VCP作为杂五原子合成子,与原位生成的取代乙烯酮反应,获得七元环内酯衍生物。
烯酮作为有机合成中常用的中间体,常用于有机催化[4]和Lewis酸催化[5]的[n+2]环加成反应(n=2-4)。最近,Louie课题组和Lu课题组发现[6],Ni和Pd配合物是烯酮环加成反应的有效催化剂,尽管烯酮在上述催化剂存在下易于分解。作者发现,通过光诱导α-重氮酮的Wolff重排(生成烯酮)有助于解决这一问题[7]。本文中,作者发展出一种新的通过可见光引发的Pd催化剂促进的α-重氮酮与VCP的不对称[5+2]环加成方法学(Scheme 1B)。该反应的关键之处在于,VCP分子反应方式的改变:Pd配位的偶极中间体II为氧杂-1,5-偶极子 (oxo-1,5-dipole),与原位生成的烯酮进行进一步反应。而其异构体I(全碳1,3-偶极子),则后续发生[3+n]环加成反应。尽管设计看似简单,但要实现预期的化学选择性和周边选择性仍存在一些挑战。首先,中间体II的分子内环化与分子间环加成存在强烈的竞争。第二,中间体II中的C或O位置均可与烯酮反应,则获得不同的环加成产物。第三,不对称地构建两种不同的立体中心,尤其涉及手性季碳中心的构建时,存在明显的挑战。
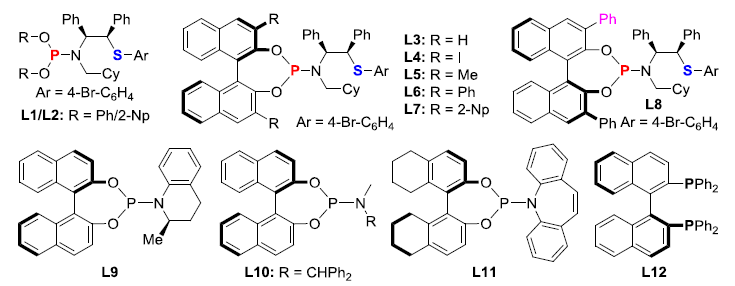
首先,作者采用乙烯基环丙烷1a’和α-重氮酮2a作为模型底物,进行了相关不对称[5+2]环加成反应条件的筛选(Table 1)。在对配体、溶剂等条件进行大量筛选后,作者发现,通过L6作为配体,Pd2dba3•CHCl3作为催化剂,丙酮作为溶剂,可在6 W蓝色LED照射以及室温条件下进行反应,获得92%收率的产物3aa(96:4 er和7:1 dr)。
在获得上述最佳反应条件之后,作者开始对1a与各类α-重氮酮2的反应进行深入研究(Table 2)。结果表明,反应的对映选择性不受苯环上电子效应的显著影响,均以70-92%的收率获得相应手性产物3aa-3af(er为93:7-98:2和dr为3:1-12.5:1)。而在苯环的间位引入取代基(如MeO和Cl)时,该反应条件同样可以兼容,从而获得手性产物3ag和3ah。同时,作者发现采用环状α-重氮酮底物2i参与反应时,可以获得具有螺四级立体中心的复杂分子3ai,产率为63%,er为87:13,dr为10:1。此外,作者同样对α-重氮酮中烷基取代基的改变进行了研究,将甲基通过乙基、正丁基、异丁基和苄基替换时,同样能够以51-74%的收率获得手性产物3aj-3am(er为96:4-99:1和dr为2:1-4:1)。同样,烷基中带有烯基、炔基和OTBS官能团时,同样具有良好的耐受性,能够获得52-80%收率的手性产物3an-3aq(er为96:4-97:3,dr为2:1-5:1)。

接下来,作者对α-重氮酮2a与一系列1,3-茚二酮衍生的取代乙烯基环丙烷(VCPs)1的反应进行了进一步研究(Table 3)。结果表明,当Rʹ具有烷基和电子多样性的芳基时,均能够与上述反应条件兼容,并以58-70%收率获得相应的产物3ba-3ea(er为91:9-97:3,dr为5:1-8:1)。同样,反应的有效性与对映选择性不受VCP中苯环上取代基电子效应的显著影响,当采用L7作为配体时,同样可以获得相应的内酯产物3fa(收率64%,er为94:6和dr为2:1)和3ga(收率53%,er为94:6和dr为5:1)。

随后,作者同样对该反应的实用性进行了研究(Scheme 2)。首先,在标准条件下,采用VCP 1a和α-重氮酮2a进行克级放大反应,与预期一致,以较高的收率获得手性环加成产物3aa。其次,以Ru 627和Ti(Oi-Pr)4作为催化剂,可以将含二烯基的化合物3ao进一步转化为结构更为复杂的多环内酯4,并且不影响其光学纯度。

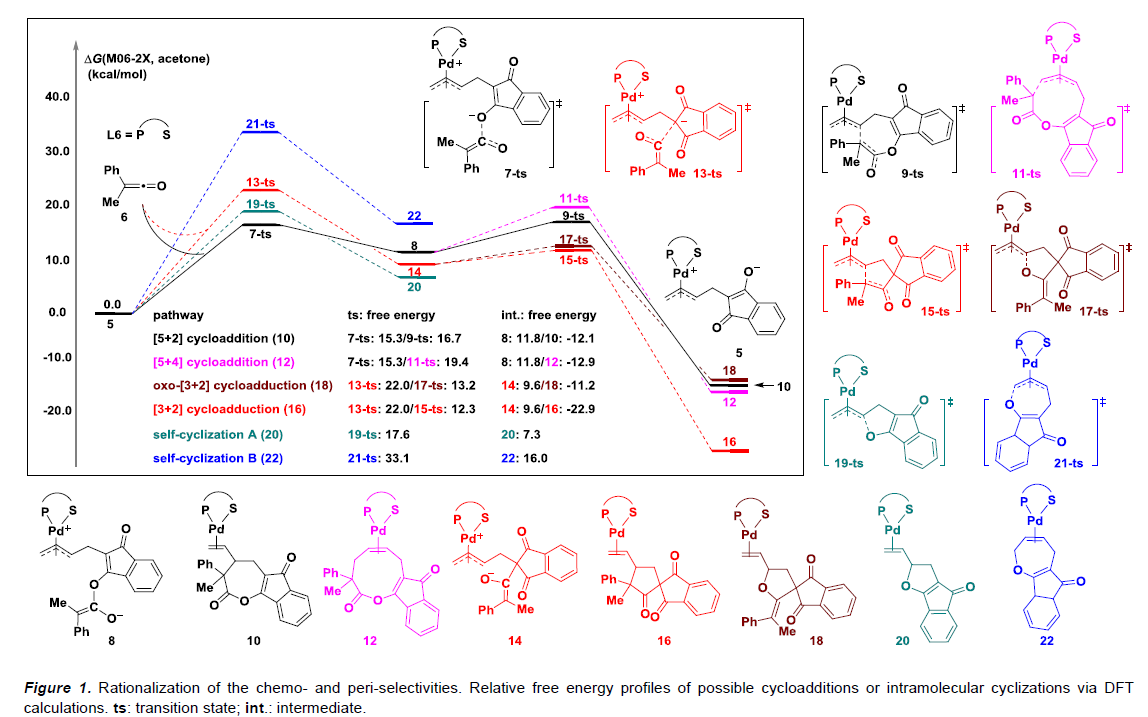
此外,为了更好地了解该反应过程中较高的化学选择性与周边选择性(periselectivity),作者使用了Gaussian 09软件进行了计算研究(Figure 1)。以常见的含Pd偶极int. 5开始,从而计算了VCP 1a与乙烯酮6的[5+2]环加成反应、竞争性环加成反应和分子内环化反应的所需的自由能。在DFT计算中,相对于中间体5和乙烯酮6的所有可能中间体和过渡态的相对吉布斯自由能均被检测。Int. 5和乙烯酮6分子间环加成比分子间环化过程放热更大,体现分子内环化过程建立于20和22的基础上,从而解释了VCP 1a’确实进行了分子内环化,同时未观察到VCP 1a的分子内环化。对比了四个可能的环加成, Int. 5的O-原子加成的过渡状态比C-原子加成所需的活化自由能低得多。此外,通过分析过渡态9-ts和11-ts之间的自由能差,七元环10比九元环12的过程更容易。这些结果表明,分子间的[5+2]环加成反应在动力学上最有利。
总结
华中师范大学陆良秋教授课题组和重庆大学蓝宇教授(理论计算)共同合作,首次实现了Pd催化条件下 可见光引发的VCP与α-重氮酮之间的不对称[5+2]环加成反应,获得23个七元内酯化合物(收率高达92%,er高达99:1,dr高达12.5:1)。该反应能够从简单易得的VCP和α-重氮酮出发,十分容易地合成出结构复杂且高度官能团化的七元内酯衍生物。此外,手性P,S-配体的设计,对于反应的选择性至关重要。同时,DFT计算研究,对反应过程中观察到的高度化学选择性与周边选择性进行了合理的解释。
参考文献
[1] For seleced examples via Rh catalysis, see: a) P. A. Wender, H. Takahashi, B. Witulski, J. Am. Chem. Soc. 1995, 117, 4720-4721; b) P. A. Wender, C. M. Barzilay, A. J. Dyckman, J. Am. Chem. Soc. 2001,123, 179-180; c) Z.-X. Yu, P. A. Wender, K. N. Houk, J. Am. Chem. Soc. 2004, 126, 9154-9155; d) P. A. Wender, L. O. Haustedt, J. Lim, J. A. Love, T. J. Williams, J.-Y. Yoon, J. Am. Chem. Soc. 2006, 128, 19, 6302-6303; e) R. Shintani, H. Nakatsu, K. Takatsu, T. Hayashi, Chem. Eur. J. 2009, 15, 8692-8694; f) C.-H. Liu, Z.-X. Yu, Angew. Chem. Int. Ed. 2017, 56, 8667-8671.[2] For selected examples, see: a) L. Jiao, S. Ye, Z.-X. Yu, J. Am. Chem. Soc. 2008, 130, 7178–7179; b) L. Jiao, M. Lin, Z.-X. Yu, J. Am. Chem. Soc. 2011, 133, 3, 447-461; c) M. Lin, G.-Y. Kang, Y.-A. Guo, Z.-X. Yu, J. Am. Chem. Soc. 2012, 134, 398-405; d) S. Bose, J. Yang, Z.-X. Yu, J. Org. Chem. 2016, 81, 6757-6765.
[3] For a pioneering work, see: a) I. Shimizu, Y. Ohashi, J. Tsuji, Tetrahedron Lett. 1985, 26, 3825-3828; for a review, see: b) J. Tsuji, Tetrahydron 1986, 42, 4361-4401.
[4] For selected reviews and a book, see: a) D. H. Paull, A. Weatherwax, T. Lectka, Tetrahedron 2009, 65, 6771-6803; b) A. D. Allen, T. T. Tidwell, Chem. Rev. 2013, 113, 7287-7342; c) R. L. Danheiser, Three Carbon-Heteroatom Bonds: Ketenes and Derivatives, Science of Synthesis, Vol. 23, Thieme Verlag KG, 2014.
[5] For selected examples on [2+2] cycloadditions, see: a) A. E. Taggi, A. M. Hafez, H. Wack, B. Young, W. J. Drury, T. Lectka, J. Am. Chem. Soc. 2000, 122, 7831-7832; b) B. L. Hodous, G. C. Fu, J. Am. Chem. Soc. 2002, 124, 1578-1579; c) J. E. Wilson, G. C. Fu, Angew. Chem. Int. Ed. 2004, 43, 6358-6360; d) C. Zhu, X. Shen, S. G. Nelson, J. Am. Chem. Soc. 2004, 128, 5352-5355; e) A. A. Ibrahim, D. Nalla, M. V.Raaphorst, N. J. Kerrigan, J. Am. Chem. Soc. 2012, 134, 2942-2945; for [2+3] cycloadditions, see: f) N. Duguet, A. M. Z. Slawin, A. D. Smith, Org. Lett. 2009, 11, 3858-3861; g) P.-L. Shao, X.-Y. Chen, S. Ye, Angew. Chem. Int. Ed. 2010, 49, 8412-8416; for [2+4] cycloadditions, see: h) T. Bekele, M. H. Shah, J. Wolfer, C. J. Abraham, A. Weatherwax, T. Lectka, J. Am. Chem. Soc. 2006, 128, 1810-1811 i) X. Xu, K. Wang, S. G. Nelson, J. Am. Chem. Soc. 2007, 129, 11690-11691; j) X.-L. Huang, L. He, P.-L. Shao, S. Ye, Angew. Chem. Int. Ed. 2009, 48, 192-195.
[6] (a) P. Kumar, D. M. Troast, R. Cella, J. Louie, J. Am. Chem. Soc. 2011, 133, 7719-7721; b) M.-M. Li, Y. Wei, J. Liu, H-W. Chen, L.-Q. Lu, W.-J. Xiao, J. Am. Chem. Soc. 2017, 139, 14707-14713; c) Y. Wei, S. Liu, M.-M. Li, Y. Li, Y. Lan, L.-Q. Lu, W.-J. Xiao, J. Am. Chem. Soc. 2019, 141, 133-137; d) J. Liu, M.-M. Li, B.-L. Qu, L.-Q. Lu, W.-J. Xiao, Chem. Commun. 2019, 55, 2031-2034; e) D. Liu, W. Ding, Q.-Q. Zhou, Y. Wei, L.-Q. Lu, W.-J. Xiao, Org. Lett. 2018, 20, 7278-7282.
[7] For a selected review on photo-Wolff rearrangement, see: a) W. Kirmse, Eur. J. Org. Chem. 2002, 2193-2256; for recent examples, see: b) Y. S. M. Vaske, M. E. Mahoney, J. P. Konopelski, D. L. Rogow, W. J. McDonald, J. Am. Chem. Soc. 2010, 132, 11379-11385; c) B. Bernardim, A. M. Hardman-Baldwinb, A. C. B. Burtoloso, RSC Adv. 2015, 5, 13311-13314; d) T. Fan, Z.-J. Zhang, Y.-C. Zhang, Jin Song, Org. Lett. 2019, 21, 19, 7897-7901; e) J. Meng, W.-W. Ding, Z.-Y. Han, Org. Lett. 2019, 21, 24, 9801-9805, and ref. 12.

















No comments yet.