
本文作者:杉杉
导读
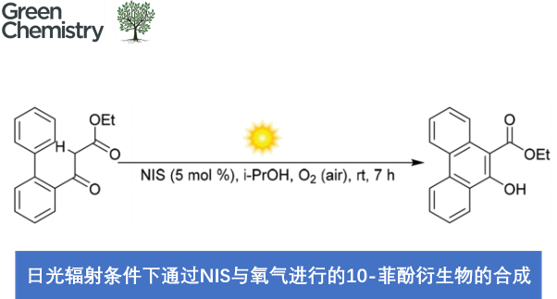
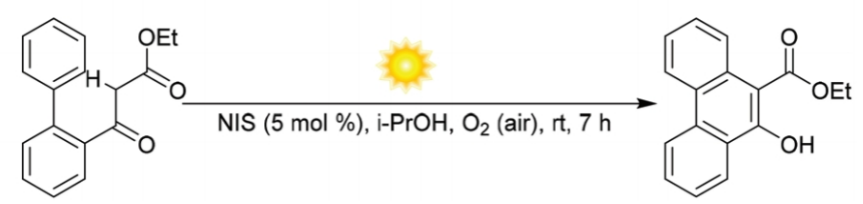
近日,中国科学院理化技术研究所的吴骊珠课题组在Green Chem.中发表论文,报道一种采用催化量的NIS与空气中O2的结合,在日光辐射以及室温反应条件下,进行的有氧氧化反应 (aerobic oxidative reaction)方法学,进而完成一系列10-菲酚衍生物 (10-phenanthrenols)的构建。同时,反应机理研究表明,原位形成的H2O2能够将I2转化为IOH,并将IOH作为后续催化循环过程中的潜在引发剂。
N-Iodosuccinimide and dioxygen in an air-enabled synthesis of 10-phenanthrenols under sunlight
J. Guo, X. Yang, B. Chen, C. Tung, L. Wu, Green Chem. ASAP. doi: 10.1039/d1gc02185k.
正文
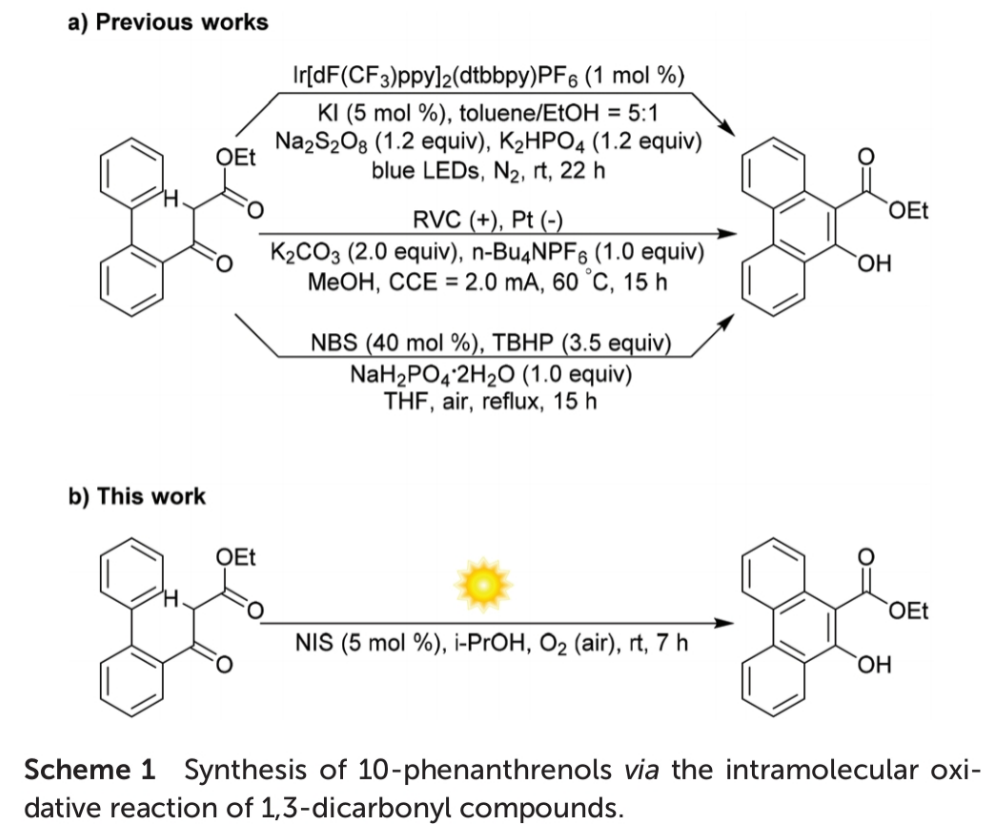
菲环 (phenanthrene) 作为基本的结构单元,广泛存在于各类生物活性天然产物、材料以及由多环芳香结构单元形成的螯合配体中。而在过去的几十年中,已经开发多种用于构建各类菲环化合物的反应策略[1]。由于具有高度的原子与步骤经济性以及起始原料易得的优势,通过选择2-联苯基取代的1,3-二羰基化合物,实现α-C(sp3)-H键直接活化的相关研究,备受有机合成化学家的关注。近期,研究表明,在化学计量的碱以及贵金属光催化剂的辅助下,通过选择光化学[2]以及 电化学[3]反应条件下,α-C(sp3)-H键断裂的反应策略,同样能够实现一系列10-菲酚衍生物的合成 (Scheme 1a)。然而,无金属试剂条件下的有氧氧化反应 (metal-free aerobic oxidation)方法学,在设计一种简洁有效与环境友好的反应策略,实现菲环骨架构建的研究中,更加具有吸引力。其中,2018年,Wang课题组[4]报道一种在过量的碱与叔丁基过氧化物存在的条件下,通过NBS诱导的分子内环芳构化 (cycloaromatization)反应策略,进而成功完成相应10-菲酚衍生物的合成 (Scheme 1a)。
此外,在可见光辐射条件下,选择碘源 (iodine source)催化,并采用O2作为终端氧化剂 (terminal oxidant)进行的氧化反应方法学[5],与选择过渡金属氧化剂促进的氧化反应方法学以及采用m-CPBA、oxone、H2O2或TBHP作为终端氧化剂参与的氧化反应方法学相比,具有更多的优势,尤其具有较低的原料成本与试剂毒性[6]。受到上述研究报道的启发,这里,本文将报道一种通过NIS引发的有氧光化学氧化分子内苯环化反应 (aerobic photo-oxidative intramolecular benzannulation)方法学,进而成功将一系列2-联苯基取代的1,3-二羰基化合物转化相应的10-菲酚类化合物 (Scheme 1b)。
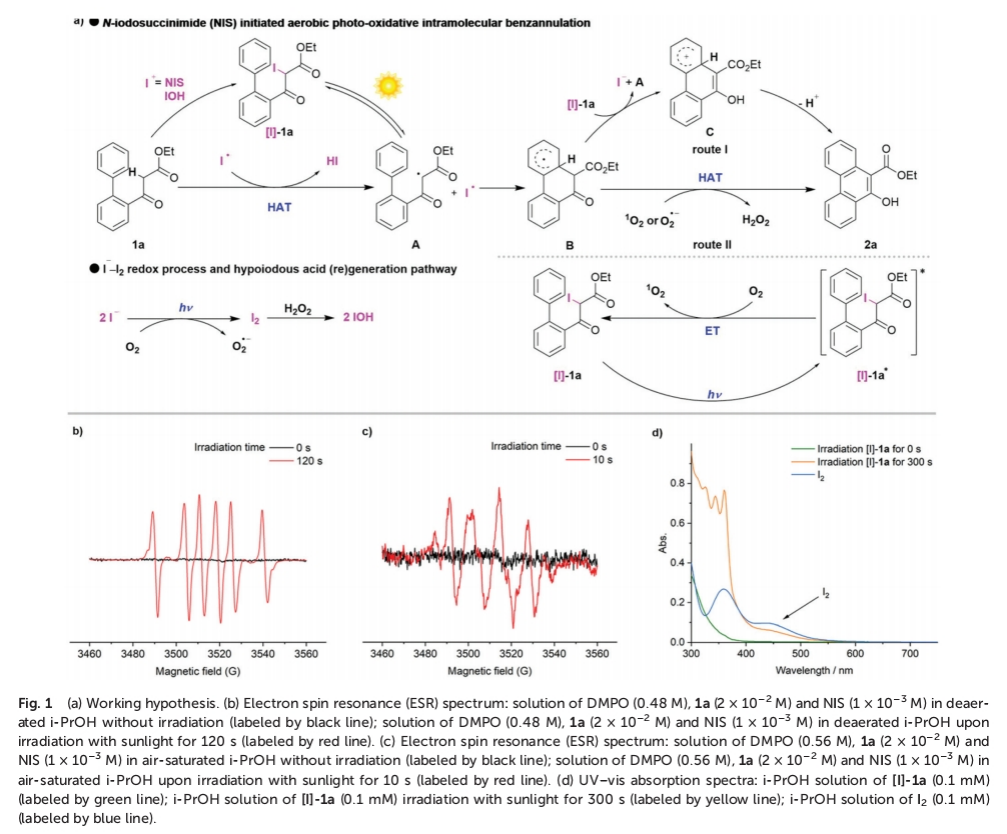
首先,作者对相关的反应机理进行深入研究 (Fig. 1)。作者发现,通过亲电试剂NIS对1a的碘化过程,形成α-碘化中间体 [I]-1a。其中,由于C(sp3)-I键的键解离能 (≈55 kcal mol–1)远低于C(sp3)-H键 (≈90 kcal mol−1),因此,在日光辐射的条件下,α-C(sp3)-I键可能发生均裂,形成α-羰基自由基 A与碘自由基 (Fig. 1a)。
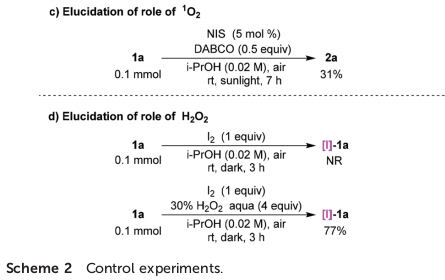
之后,该小组发现,通过EPR实验,在黑暗条件下,未检测出α-羰基自由基A的信号峰。同时,该小组观察到,将DMPO (5,5-dimethyl-1-pyrroline-N-oxide)、1a以及NIS在脱气的i-PrOH溶剂中辐射120 s后,能够观察到α-羰基自由基A与DMPO加合物的特征信号峰,进而表明α-C(sp3)-I键均裂过程的存在 (Fig. 1b)。同时,形成的碘自由基能够攫取底物1a中的一个氢原子,形成α-羰基自由基A与HI。之后,α-羰基自由基A经历分子内的自由基加成过程,形成中间体B,并通过[I]-1a的进一步氧化,产生中间体C,并伴随α-羰基自由基A与I–的形成。接下来,中间体C通过去质子化步骤,获得最终的目标产物2a (route I)。此外,作者进一步发现,同样能够通过激发态物种 (excited species)[I]-1a*经历相应的能量转移过程,形成单线态氧 (1O2),并伴随[I]-1a中间体的再生。事实上,通过高能的紫外或可见光对I–与O2体系的辐射,能够释放出O2•-与I2 (Fig. 1c与 d)。由此,中间体B能够通过1O2或O2•-进行直接的氢原子转移过程,进而原位形成产物2a与H2O2 (route II)。同时,I2能够通过H2O2的进一步氧化,形成反应活性中间体IOH。之后,通过IOH与1a作用,形成[I]-1a,并进一步参与后续的催化循环过程。
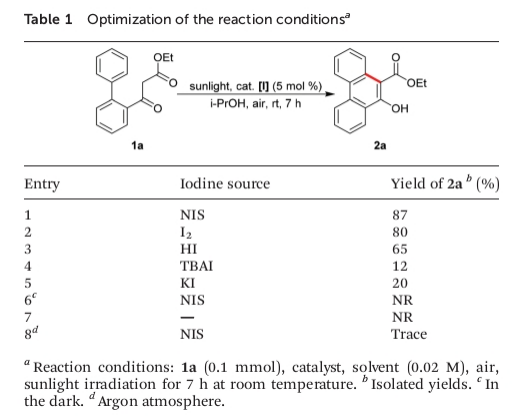
为进一步证实上述反应机理假设的合理性,作者采用1a作为模型底物,进行了相关氧化反应条件的优化筛选 (Table 1)。确定最佳的反应条件为:采用5 mol%的NIS作为催化剂,i-PrOH作为反应溶剂,在空气以及室温与日光辐射的条件下进行反应,最终获得87%收率的目标产物2a。
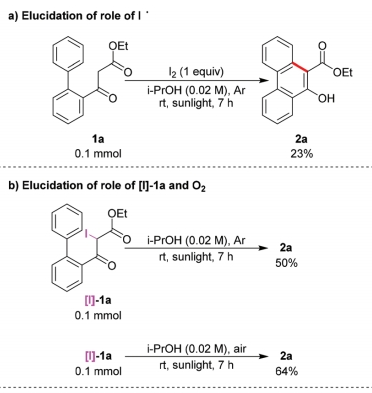
之后,作者进行一系列相关的控制实验研究 (Scheme 2)。首先,作者发现,通过碘与1a在日光辐射、脱气异丙醇溶剂以及氩气存在的条件下反应,能够获得23%收率的产物2a,进而表明碘自由基能够攫取1a中的氢原子,产生α-羰基自由基A (Scheme 2a)。之后,该小组发现,采用[I]-1a作为反应底物,在无催化剂与日光辐射以及氩气气氛存在的条件下反应,则获得50%收率的产物2a。同时,作者观察到,将上述转化过程在空气气氛中进行时,反应收率略有提高。进而表明[I]-1a与O2均能够作为上述反应过程中的氧化剂 (Scheme 2b)。此外,作者进一步发现,向1a的标准反应体系中加入1O2淬灭剂DABCO时,能够较为显著地抑制相应氧化过程的进行 (Scheme 2c)。尽管1a在无光照的条件下进行反应时,未能形成相应的中间体[I]-1a,然而,作者观察到,向反应体系中加入H2O2时,却能够获得77%收率的 [I]-1a (Scheme 2d)。
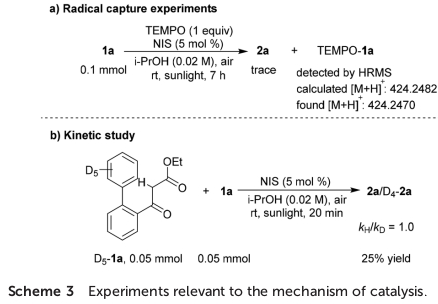
同时,该小组发现,向1a的标准反应体系中加入自由基捕获剂TEMPO时,上述的反应过程受到完全抑制,仅获得痕量的目标产物2a,并且,通过HRMS能够检测出TEMPO-1a加合物的产生。进而进一步支持在日光辐射条件下,通过[I]-1a中α-C(sp3)-I键的断裂,形成α-羰基自由基A与碘自由基的机理步骤 (Scheme 3a)。此外,作者在通过竞争实验进行的KIE研究中,由相关产物的1H-NMR分析,最终确定kH/kD速率比为1。进而表明在反应机理的决速步骤中,未涉及去质子化过程 (Scheme 3b)。
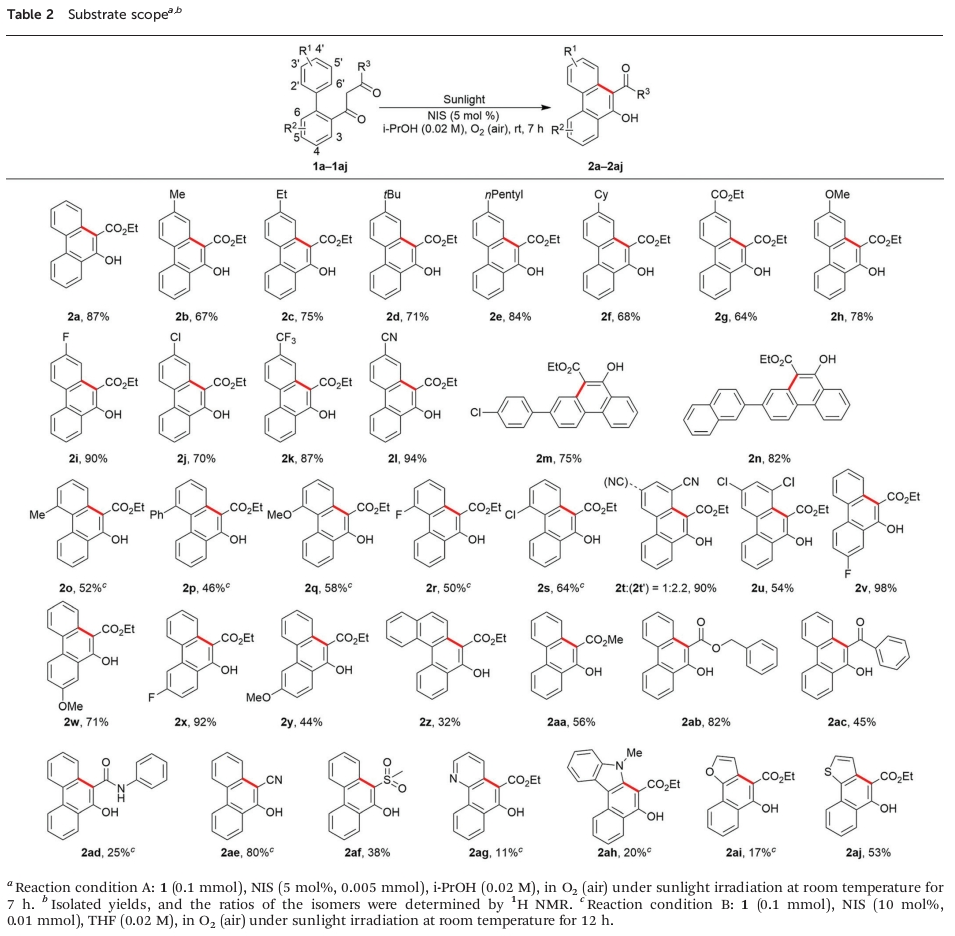
在理解相关的反应机理之后,作者开始对相关底物的应用范围进行考察 (Table 2)。研究表明,在芳环中的4′-位置具有供电子与吸电子基团取代时,均能够较好地与上述的最佳反应条件兼容,并获得相应的目标产物2b–2n,收率为64-94%。在Condition B的条件下,对于芳环中的2′-位置具有立体位阻的供电子与吸电子基团取代的底物,同样能够良好地参与上述的苯环化过程,并获得相应目标产物2o–2s,收率为46-64%。同时,上述的标准反应条件对于在芳环中的3′-位置具有氰基取代的底物,则能够获得90%收率的2t与2t’的混合物,产物为1:2.2。而对于在芳中环的3′-/5′-位置同时具有氯取代基团的底物,最终能够获得54%收率的苯环化产物2u。
之后,作者进一步发现,R2为氟基团取代时,能够获得相应的目标产物2v (98%收率)与2x (92% 收率)。然而,R2为甲氧基取代时,反应收率出现显著降低,并获得产物2w (71% 收率)与2y (44% 收率)。同时,多环芳香底物1z同样能够与上述的苯环化反应条件良好地兼容,并获得32% 收率的目标产物2z。此外,R3为-COOMe、-COOBn、-COPh、-CONHPh、-CN以及-SO2Me基团取代时,能够获得相应的苯环化产物2aa–2af,收率为25-82%。接下来,作者发现,上述的最佳反应条件对于杂环芳基,例如吡啶、吲哚、呋喃或噻吩取代的相应底物,则仅能够获得较低至中等反应收率的苯环化产物2ag–2aj。
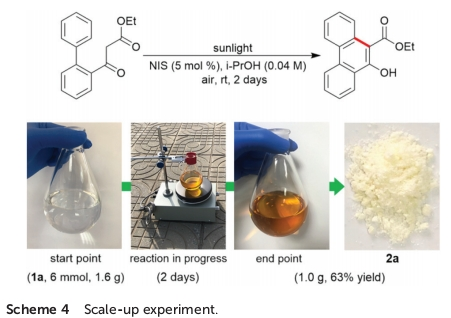
最后,作者发现,将底物1a的用量扩大至6 mmol时,同样能够获得63%收率的苯环化产物2a,进而表明这一全新的苯环化策略具有较好的工业应用价值 (Scheme 4)。
总结
中国科学院理化技术研究所的吴骊珠课题组报道一种较为清洁与绿色的,并通过日光驱动的C(sp3)-H键活化策略,进而成功实现一系列10-菲酚衍生物的合成。反应过程中选择催化量的NIS,并采用O2作为终端氧化剂,同时无需选择金属试剂以及强氧化剂。反应机理研究表明,通过I–-I2氧化还原体系参与的反应过程,能够在有氧的光化学氧化条件下进行,进而产生O2•−作为活性氧物种,并参与后续的氢原子转移过程,原位形成H2O2。之后,通过H2O2对I2的氧化,形成潜在的引发剂IOH。此外,这一全新的苯环化策略具有高度的原子与步骤经济性、底物应用范围广泛、高度的官能团兼容性以及反应条件温和等优势。
参考文献
[1] (a) C. C. McAtee, P. S. Riehl, C. S. Schindler, J. Am. Chem. Soc. 2017, 139, 2960. doi: 10.1021/jacs.7b01114.(b) T. Matsushima, S. Kobayashi, S. Watanabe, J. Org. Chem. 2016, 81, 7799. doi: 10.1021/acs.joc.6b01450.
[2] Q. Teng, L. Xu, D. Cheng, X. Xu, Chin. J. Org. Chem. 2020, 40, 4258. doi: 10.6023/cjoc202005077. [3] R. Mei, C. Yang, F. Xiong, M. Mao, H. Li, J. Sun, L. Zou, W. Ma, L. Ackermann, Adv. Synth. Catal. 2021, 363,1120. doi: 10.1002/adsc.202001431. [4] Y. T.Jiang, Z. Z.Yu, Y. K. Zhang, B. Wang, Org. Lett. 2018, 20, 3728. doi: 10.1021/acs.orglett.8b01160. [5] (a) N. A. Romero, D. A. Nicewicz, Chem. Rev. 2016, 116, 10075. doi: 10.1021/acs.chemrev.6b00057.(b) J. Chen, X. Hu, L. Lu, W. Xiao, Chem. Soc. Rev. 2016, 45, 2044. doi: 10.1039/C5CS00655D.
(c) C. K. Prier, D. A. Rankic, D. W. C. MacMillan, Chem. Rev. 2013, 113, 5322. doi: 10.1021/cr300503r.
(d) D. A. Nicewicz, D. W. C. MacMillan, Science 2008, 322, 77. doi: 10.1126/science.1161976.
[6] (a) R. Wang, L. Wang, Q. Xu, B. Ren, F. Liang, Org. Lett. 2019, 21, 3072. doi: 10.1021/acs.orglett.9b00655.(b) S. Liu, R. Wang, B. Zhu, W. Guan, F. Liang, Org. Biomol. Chem. 2019, 17, 8695. doi: 10.1039/C9OB01520E.
(c) P. T. Parvatkar, R. Manetsch, B. K. Banik, Chem. – Asian J. 2019, 14, 6. doi: 10.1002/asia.201801237.
(d) B. Chen, L. Wu, C. Tung, Acc. Chem. Res. 2018, 51, 2512. doi: 10.1021/acs.accounts.8b00267.
(e) Y. Liu, B. Wang, X. Qiao, C.-H. Tung, Y. Wang, ACS Catal. 2017, 7, 4093. doi: 10.1021/acscatal.7b00799.
(f) D. Staveness, I. Bosque, C. R. J. Stephenson, Acc. Chem. Res. 2016, 49, 2295. doi: 10.1021/acs.accounts.6b00270.
(g) J. Chen, X. Hu, L. Lu, W. Xiao, Acc. Chem. Res. 2016, 49, 1911. doi: 10.1021/acs.accounts.6b00254.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!

















No comments yet.