

本文作者:杉杉
导读
近日,Washington大学的C. K. Luscombe课题组在Org. Lett.中发表论文,报道一种在室温条件下进行的苯并呋喃化合物的直接C-H芳基化反应方法学。这一全新的芳基化策略具有优良的C-2区域选择性,并选择市售的芳基碘作为芳基源,同时,具有良好的官能团兼容性。反应机理研究表明,通过芳基化过程形成的Heck型氧芳基化 (oxyarylation)产物,能够作为钯碳化 (carbopalladation)中间体存在的关键证据。
Room Temperature C-H Arylation of Benzofurans by Aryl Iodides
A. L. Mayhugh, C. K. Luscombe, Org. Lett. ASAP doi: 10.1021/acs.orglett.1c02397.

正文
苯并呋喃及其衍生物作为重要的结构单元,广泛存在于药物以及材料等分子中。其中,α-芳基化反应方法学是构建苯并呋喃骨架的重要策略。同时,传统的交叉偶联反应策略,目前仍然应用较为广泛。此外,苯并呋喃底物的直接C-H芳基化策略已经开始受到合成化学家的关注。然而,上述C-H芳基化策略中却存在反应温度过高以及热敏性官能团无法有效兼容的问题,从而使这一策略的进一步应用受到较大限制[1]。尽管少数较为温和的C-H芳基化策略,目前已经有相关的文献报道[2],然而,同样存在室温条件下反应速率缓慢、底物应用范围较为有限以及采用易于分解并难以获得的芳香重氮盐等局限。与此同时,尽管涉及杂环联芳基化合物 (heterobiaryl)构建的室温芳基化反应方法学,已有相关的研究报道,例如吲哚以及苯并噻吩化合物的C-H芳基化反应方法学[3]。然而,实现苯并呋喃化合物的室温直接芳基化反应,则仍有待进一步研究。为解决这一问题,本文作者报道一种全新的采用芳基碘作为芳基源,进行的苯并呋喃化合物的C-2区域选择性芳基化反应方法学。
首先,作者采用苯并呋喃化合物1a与碘苯2a作为模型底物,进行了相关反应条件的优化筛选 (Scheme 1)。进而确定最佳的反应条件为:采用Pd(OAc)2作为催化剂,Ag2O作为银盐前体,2-硝基苯甲酸作为添加剂,在六氟异丙醇 (HFIP)溶剂中,室温条件下进行反应,最终获得90%收率的偶联C-H芳基化产物3a。

在上述的最佳反应条件下,作者首先对芳基碘底物的应用范围进行考察 (Scheme 2)。研究表明,一系列芳基不同位置带有供电子与吸电子基团取代的芳基碘底物,均能够较好地与上述的最佳反应条件兼容,并以35-94%收率获得相应的C-H芳基化产物3a–3o。值得注意的是,这一全新的C-H芳基化策略具有良好的官能团兼容性,对于芳基碘底物中存在的卤素取代基、醇羟基、醛基以及酯基等,均能够有效地进行兼容。更为有趣的是,在选择2-碘苯酚作为反应底物时,能够进行进一步的串联反应过程,并以较高的反应收率获得相应的4b,9b-二氢苯并呋喃[3,2-b]苯并呋喃产物 (4)。

接下来,作者进一步对苯并呋喃底物的应用范围进行考察 (Scheme 3)。研究表明,在苯并呋喃底物中的芳基不同位置具有异丙基、溴、甲氧基以及硼酸酯基团取代时,均能够顺利地参与上述的C-H芳基化过程,并以41-74%的收率,获得相应的目标产物3p–3s。同时,该小组发现,上述的最佳反应条件对于C-3位带有甲基取代的苯并呋喃底物,同样能够良好地兼容,并以82%的反应收率获得相应C-H芳基化产物3t。值得注意的是,这一全新的C-H芳基化策略同样能够应用于皮肤病治疗药物xanthotoxin衍生物3u的合成。然而,作者进一步观察到,将上述的标准反应条件应用于双甲基取代的呋喃底物时,则未能获得相应的目标产物3v。

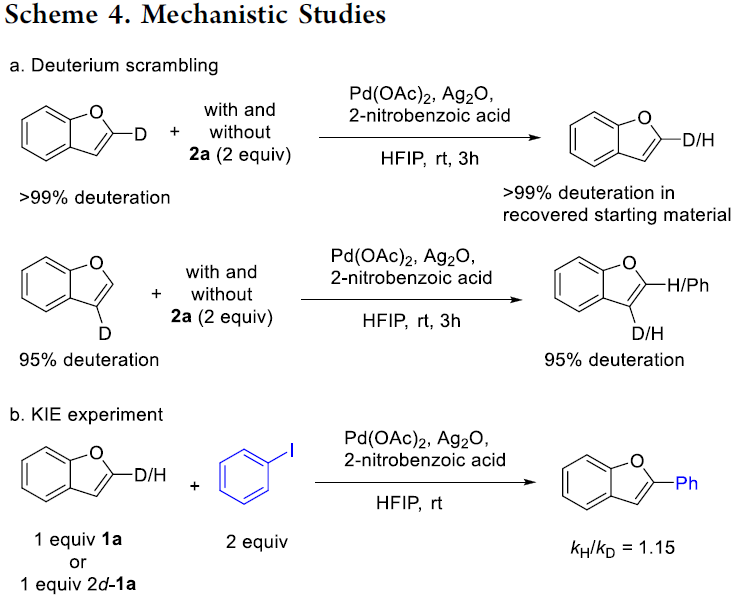
为阐明合理的反应机理,作者进行一系列相关的实验研究 (Scheme 4)。首先,该小组在上述标准反应体系中存在2a或无2a存在时,分别与2d–1a以及3d–1a进行相关的氢氘交换实验研究 (Scheme 4a)。实验中作者观察到,与通过协同金属化-去质子化 (CMD)路径进行的Pd或Ag媒介的C-H活化过程的预测相反,在上述的实验条件下,均未观察到相应的氢氘置乱现象。而在反应过程通过CMD机理进行的情况下,如果C-H活化过程为决速步骤,同时反应过程可逆,则能够在C-2位置观察到相应的氢氘置乱现象。并且,通过氢氘交换实验的研究,同样能够进一步排除非决速的C-H活化过程以及可逆的CMD机理[4]-[5]。
之后,为进一步研究反应机理过程中的驱动因素,作者对苯并呋喃C-2位的KIE进行深入研究 (Scheme 4b)。实验观察到C-2位的SKIE (secondary KIE)数值为1.15,进而表明,上述的C-H芳基化过程无法通过C-H活化作为决速步骤的CMD路径进行。除CMD路径之外,Pd催化C-H芳基化反应过程同样涉及其它的机理路径,例如Heck型机理[6]以及亲电芳香取代 (SEAr)机理[7]。其中,苯并呋喃的SEAr反应过程,能够选择性地在C-2位置进行,这与亲电硝化以及亲电酰化过程中所观察到的区域选择性一致。并且C-2位置的选择性,通过具有稳定化苄基碳正离子特性的σ-配合物中间体进行驱动,进而实现芳基化过程中预期的区域选择性。然而,C-2位置观察到的正的SKIE数值,并无法表明在C-2位置中存在相关的钯化 (palladation)过程。即由于去质子化速率与芳基正离子 (arenium ion)的形成速率相当,因此,通常情况下,芳香亲电取代过程的KIE数值较小,甚至无法观察到KIE的存在。由此,作者假设,C-2位的钯化过程,仍然可能通过SEAr机理进行。
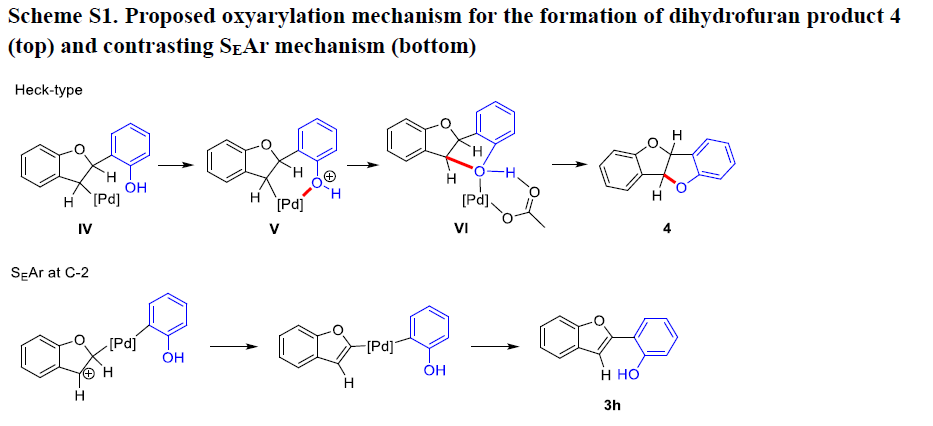
接下来,该小组通过2-碘苯酚环化,形成产物4的反应过程,进而证实具有sp3碳中心的C-3-钯配合物的存在。并且,产物4的形成过程,与预期的Heck型氧芳基化过程相一致。其中,邻近的羟基能够进一步取代苯并呋喃环中的钯基团 (Scheme S1)。相反地,通过SEAr机理进行的C-2钯化过程中,在C-3位置能够形成相应的碳正离子中心 (Scheme S1)。同时,在酸性条件下,2-碘苯酚中的羟基官能团无法与碳正离子中心进行成键。之后,作者考虑到,同样可能存在一种与芳基化过程相互独立的二级C-O键的形成过程。而且,即使在底物完全转化之前,将反应过程停止,同样无法获得相应的芳基化产物 (3h)。并且,对于其他未保护的醇底物,在上述标准反应条件下的稳定性的相关研究,同样无法证实SEAr过程在C-O环化步骤之前进行。同时,上述的实验观察与Pd催化的烯基化合物的氧芳基化/C-H官能团化过程一致,进而表明这一全新的C-H芳基化过程与Heck化学之间具有相似性。而且,通过上述实验观察,能够进一步排除C-2位芳基化过程通过SEAr机理进行的可能性。
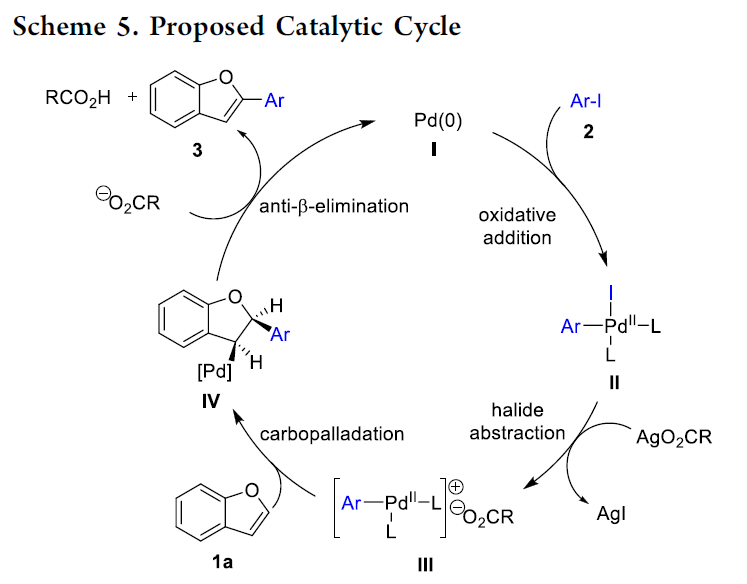
最后,结合上述的实验研究,作者假设这一全新的C-H芳基化过程通过Heck型机理路径进行 (Scheme 5)。首先,通过芳基碘与Pd(0)之间的氧化加成过程,形成中间体II,之后,中间体II与原位形成的羧酸银盐之间通过卤离子攫取 (halide abstraction)步骤,形成具有更高反应活性的钯金属中心,同时,通过钯金属中心中新引入的羧基负离子配体的进一步解离,形成钯阳离子配合物 III。接下来,1a与钯阳离子配合物 III之间经历钯碳化 (carbopalladation)步骤,形成中间体IV,并进一步通过碱辅助的anti-β-消除步骤,进而获得目标产物3,同时,使Pd(0)催化剂再生。之后,作者进一步假设,卤离子攫取以及后续的配体解离过程,是上述C-H芳基化反应过程能够在室温条件下顺利进行的关键。此外,除通过氢键作用[8],使活性钯配合物稳定化之外,HFIP同样可能与羧酸银之间进行相应的酸碱交换过程[9]。
总结
Washington大学的C. K. Luscombe课题组报道一种全新的苯并呋喃衍生物的区域选择性C-2芳基化反应方法学。并且,这一全新的C-H芳基化策略具有反应条件温和以及极为优良的官能团兼容性等优势。同时,作者设想,上述的C-H芳基化过程通过Heck型芳基化机理进行。
参考文献
[1] (a) S. C. Yin, Q. Zhou, X. Y. Zhao, L. X. Shao, J. Org. Chem. 2015, 80, 891. doi: 10.1021/acs.joc.5b01544.(b) J. Cao, Z. L. Chen, S. M. Li, G. F. Zhu, Y. Y. Yang, C. Wang, W. Z. Chen, J. T. Wang, J. Q. Zhang, L. Tang, Eur. J. Org. Chem. 2018, 22, 2774. doi: 10.1002/ejoc.201800374.
(c) S. Mao, X. Shi, J. F. Soulé, H. Doucet, Eur. J. Org. Chem. 2020, 2020, 91. doi: 10.1002/ejoc.201901561.
[2] (a) H. P. L. Gemoets, I. Kalvet, A. V. Nyuchev, N. Erdmann, V. Hessel, F. Schoenebeck, T. Noël, Chem. Sci. 2017, 8, 1046. doi: 10.1039/C6SC02595A.(b) A. F. P. Biajoli, E. T. Da Penha, C. R. D. Correia, RSC Adv. 2012, 2, 11930. doi: 10.1039/C2RA22213B.
(c) Z. Xu, Y. Xu, H. Lu, T. Yang, X. Lin, L. Shao, F. Ren, Tetrahedron 2015, 71, 2616. doi: 10.1016/j.tet.2015.03.051.
[3] (a) N. Lebrasseur, I. Larrosa, J. Am. Chem. Soc. 2008, 130, 2926. doi: 10.1021/ja710731a.(b) C. Colletto, A. Panigrahi, J. Fernández-Casado, I. Larrosa, J. Am. Chem. Soc. 2018, 140, 9638. doi: 10.1021/jacs.8b05361.
[4] S. I. Gorelsky, D. Lapointe, K. Fagnou, J. Org. Chem. 2012, 77, 658. doi: 10.1021/jo202342q. [5] M. D. Lotz, N. M. Camasso, A. J. Canty, M. S. Sanford, Organometallics 2017, 36, 165. doi: 10.1021/acs.organomet.6b00437. [6] C. Colletto, S. Islam, F. Juliá-Hernández, I. Larrosa, J. Am. Chem. Soc. 2016, 138, 1677. doi: 10.1021/jacs.5b12242. [7] S. I. Gorelsky, D. Lapointe, K. Fagnou, J. Org. Chem. 2012, 77, 658. doi: 10.1021/jo202342q. [8] T. Bhattacharya, A. Ghosh, D. Maiti, Chem. Sci. 2021, 12, 3857. doi: 10.1039/D0SC06937J. [9] C. Colletto, S. Islam, F. Juliá-Hernández, I. Larrosa, J. Am. Chem. Soc. 2016, 138, 1677. doi: 10.1021/jacs.5b12242.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!














No comments yet.