
本文作者:杉杉
导读
锍盐因其较为独特的分子结构与高度的化学反应活性,长期以来,备受合成化学家的广泛关注。并且,锍盐类化合物能够较为容易地进行相应的单电子还原过程,进而能够更为有效地形成有机合成中较为关键的芳基自由基中间体。然而,通过锍盐形成不稳定的烷基自由基则具有较高的挑战性。近日,南京大学史壮志与陆红健课题组报道通过S-(烷基)噻蒽鎓盐形成的不稳定烷基自由基作为关键中间体,进而在较为温和的光氧化还原条件下,实现多种不同类型的合成转化过程,并表现出良好的可控性与选择性。同时,上述策略的发展,为C-OH键向C-B与C-C键的转化开辟出全新的路线。
Generation of non-stabilized alkyl radicals fromthianthrenium salts for C-B and C-C bondformation
C. Chen, Z. Wang, H. Lu, Y. Zhao, Z. Shi, Nat. Commun. ASAP. doi: 10.1038/s41467-021-24716-2.
正文
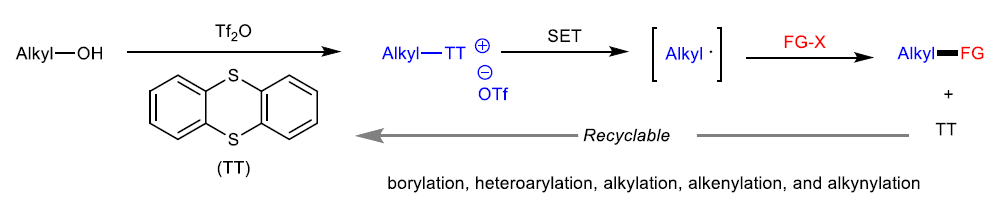
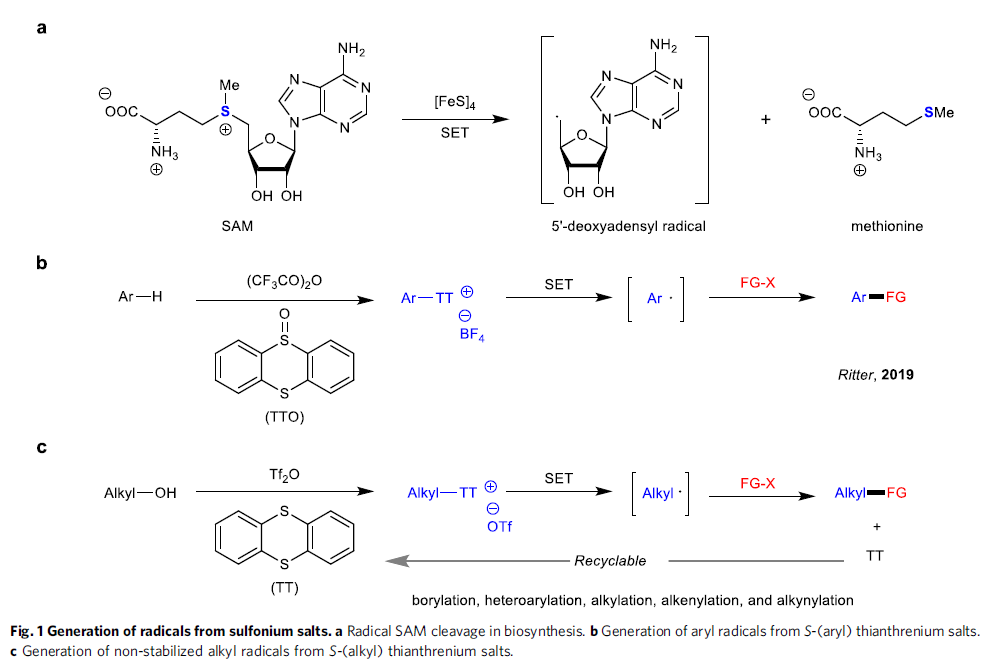
锍盐已经成为有机合成研究中应用最为广泛的活性中间体之一。通常,锍盐的反应活性取决于其硫原子中带有的正电荷。基于上述理论,目前已经成功设计出诸多的锍盐基试剂 (sulfonium salt-based reagent),例如Umemoto试剂,并将其进一步应用于亲电取代反应方法学的研究[1]-[3]。迄今为止,天然存在的,能够促进锍盐形成烷基自由基的酶,均归属于自由基S-腺苷甲硫氨酸(S-adenosylmethionine, SAM)体系[4]-[5](Fig. 1a)。在过去几十年中,已经成功设计出采用锍盐作为自由基前体的合成转化策略[6]-[7]。近年来,光氧化还原催化作为有机合成研究中的新兴领域,为SET诱导合成转化策略的研究,开辟出全新的设计思路。Ritter课题组[8]-[9]报道一系列芳香底物的位置选择性C-H官能团化反应方法学,进而成功完成各类S-(芳基)噻蒽鎓盐分子的构建,并将其进一步应用于采用光氧化还原催化的,通过自由基中间体进行的相关合成转化过程的研究(Fig. 1b)。同时,Procter课题组[10]进一步设计出一种采用有机光氧化还原催化剂,并通过S-(芳基)二苯并噻吩鎓盐 (S-(aryl) dibenzothiophenium salt)中间体,进一步构建(杂)联芳类化合物的一锅反应策略。然而,通过锍盐参与的SET过程,通常仅局限于形成相应的芳基自由基。而通过锍盐形成烷基自由基,尤其较不稳定的烷基自由基,则存在巨大挑战。
作为S-叶立德的前体,S-(烷基)锍盐已经广泛应用于环化与重排反应方法学的相关研究[11]-[12]。在早期的研究中,Kellogg课题组成功设计出在光氧化还原催化条件下,S-(烷基)锍盐的还原反应方法学[13]-[14]。然而,目前为止,相关的文献报道仅限于形成较为稳定的烷基自由基,例如苄基或具有α-吸电子基团的烷基自由基。由此,南京大学史壮志与陆红健课题组设计出一系列通过S-(烷基)噻蒽鎓盐形成的不稳定烷基自由基中间体,进行的C-B与C-C键构建的合成转化策略(Fig. 1c)。
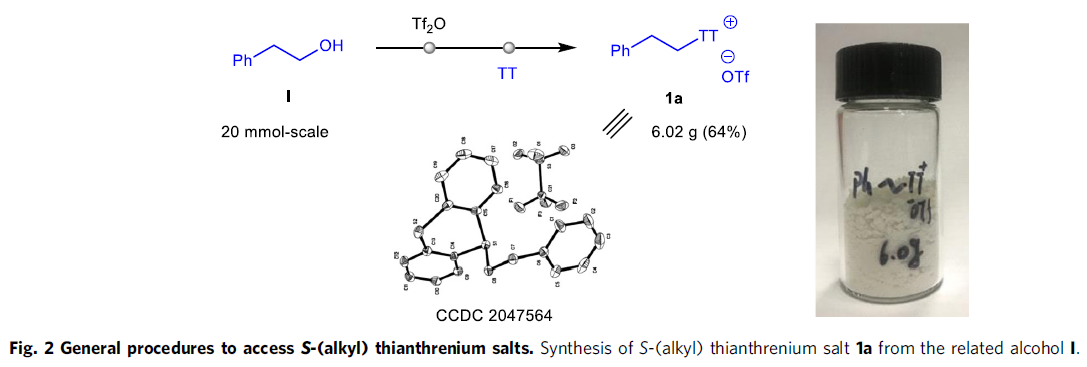
S-(烷基)噻蒽鎓盐的合成(Fig. 2)。作者通过相应的醇原料与TT (thianthrene)以及Tf2O试剂的一锅反应策略,替代上世纪90年代采用的具有极高毒性的烷基汞与烷基锡试剂[15]-[16],进而顺利完成各类S-(烷基)噻蒽鎓盐的克级规模合成(例如1a, Fig. 2)。
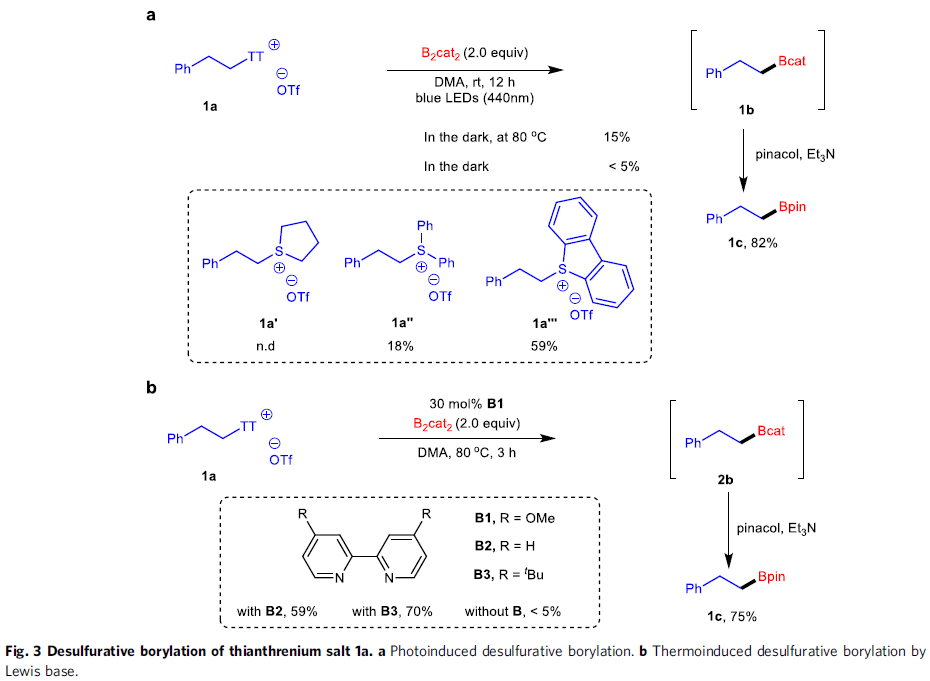
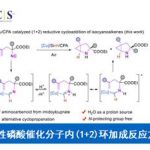
反应设计(Fig. 3)。烷基硼酸酯作为有机合成中的关键砌块,能够进一步转化为多种不同类型的官能团化有机分子。传统烷基硼酸酯的合成方法主要涉及采用有机锂或Grignard试剂参与的亲电硼化 (electrophilicborylation)方法学以及烯基化合物的硼氢化反应方法学。近期,则更多地选择较为稳定的烷基自由基前体[17]-[22],尤其是具有氧化还原活性的烷基卤、N-羟基邻苯二甲酰亚胺(N-hydroxyphthalimide, NHPI)酯以及Katritzky盐等,进而完成一系列烷基硼酸酯的制备。鉴于烷基硼酸酯在有机化学研究中的重要价值,作者开始探索在光以及热诱导条件下,噻蒽鎓盐参与的去硫硼化反应 (desulfurativeborylation)方法学(Fig. 3)。研究表明,在蓝光LED辐射条件下,1a与2.0eq.B2cat2(bis(catecholato)diboron)在DMA溶剂中进行反应,形成中间体1b,之后,在三乙胺存在下,进一步与频哪醇作用,转化为更加稳定的硼酸酯1c,收率为82%。同时,作者发现,采用其它锍盐,例如,锍盐1a′则无法有效地参与相应的去硫硼化过程;锍盐1a′′,最终获得PhBpin作为主要产物;锍盐1a′′′,尽管反应活性较低,然而,却能够获得中等收率的目标产物。并且,近期,本课题组已经成功开发出采用Lewis碱促进的,通过Katritzky盐参与的去氨基硼化反应 (deaminativeborylation)方法学[23]。受到上述研究的启发,作者进一步设计出通过Lewis碱促进的热诱导去硫硼化反应策略。例如,在Lewis碱4,4′-二甲氧基-2,2′-联吡啶(B1)存在下,将1a与B2cat2在DMA溶剂中,80 oC下进行加热,同样能够获得75%收率的预期产物1c。同时,作者发现,其他Lewis碱,例如2,2′-联吡啶(B2)以及4,4′-二叔丁基-2,2′-联吡啶(B3)则均表现出较低的反应活性。
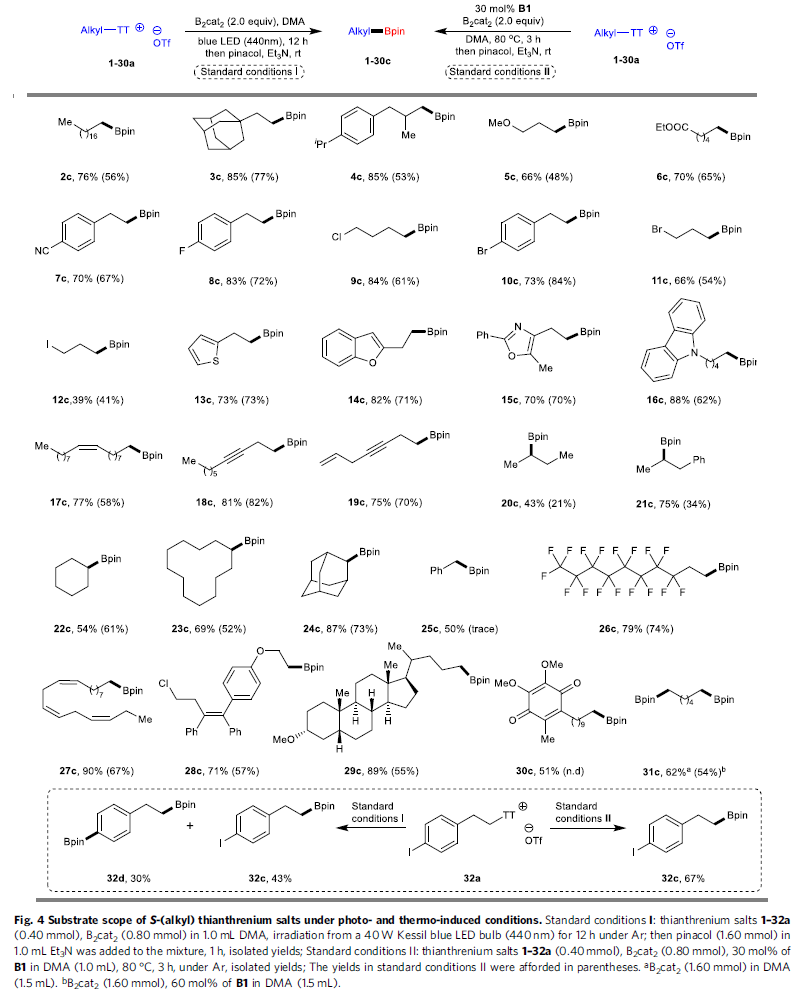
去硫硼化方法学的底物应用范围(Fig. 4)。在上述的标准反应条件下,作者进一步考察相应的光诱导与热诱导去硫硼化反应方法学中,噻蒽鎓盐的底物应用范围。研究表明,一系列一级与二级烷基噻蒽鎓盐底物,在上述的标准条件下,均能够与B2cat2顺利地进行相应的硼化过程,并获得目标产物2c–24c,收率为34-88%。同时,作者发现,苄基噻蒽鎓盐25a同样能够与相应光诱导的反应条件良好地兼容。然而,在热诱导条件下,则仅能够观察到痕量的预期产物25c。并且,具有全氟烷基碳链的噻蒽鎓盐底物26a,同样能够较为容易地进行相应的硼化过程,并获得目标产物26c。而且,具有三重顺式双键的α-亚麻酸 (linolenic acid)衍生的噻蒽鎓盐底物27a,同样能够顺利完成上述的去硫硼化过程,并形成双键构型完全保持的硼化产物。同时,作者选择结构更为复杂的底物28a–30a,深入考察上述两种反应体系中的官能团兼容性。该小组发现,idebenone衍生物30a,在可见光辐射条件下,能够以中等程度的反应收率获得相应产物30c,然而,在Lewis碱体系中,则无法有效地参与上述的去硫硼化过程。此外,该小组进一步发现,具有双重锍盐结构单元的底物31a,同样能够与上述的热诱导与光诱导去硫硼化反应体系良好地兼容。值得注意的是,在多数的反应实例中,光诱导过程能够表现出更为优良的反应活性。同时,作者进一步发现,芳基中具有C-I键的噻蒽鎓盐底物(32a),却在热反应条件下,表现出更加优良的化学选择性。而光化学反应条件下,则无法获得良好的化学选择性。
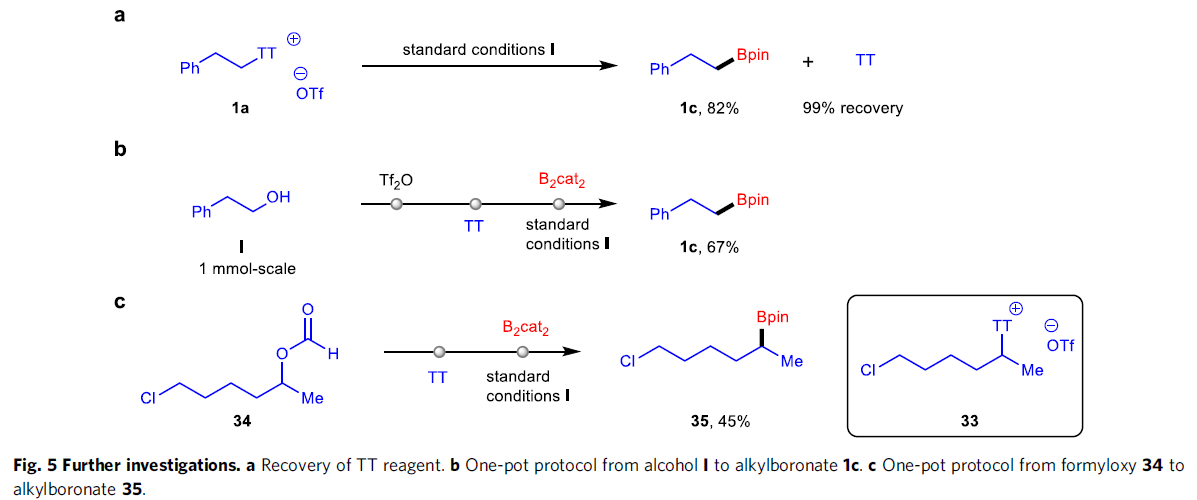
合成应用研究(Fig. 5)。首先,作者发现,1a在上述标准条件下,能够形成TT试剂与主要产物1c。并且,将上述反应混合物采用硅胶快速柱色谱进行分离纯化之后,能够以良好的反应收率获得目标产物1c,并以近乎定量的回收率,使TT试剂再生(Fig. 5a)。为进一步阐明上述去硫硼化方法学在简化合成路线方面的应用潜力,接下来,作者尝试相应一锅反应的研究。例如,首先采用醇化合物I进行一锅磺化与噻蒽化 (thianthrenation),之后,在蓝光辐射条件下,进行后续的硼化反应过程,能够以67%的收率形成烷基硼酸酯1c(Fig. 5b)。前期已有文献研究表明,带有氯基团的二级烷基噻蒽鎓盐33通常较不稳定,并难以制备。然而,这里作者通过一锅反应的策略设计,却能够较为便捷地将34中的甲酰氧基转化为带有氯基团的产物35(Fig. 5c)。
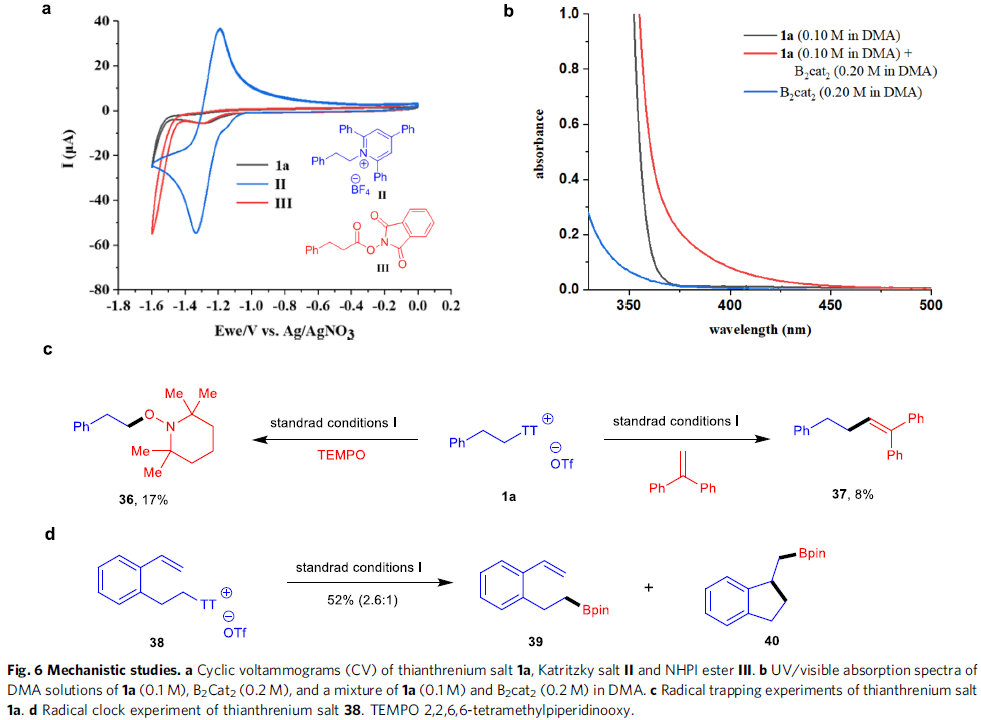
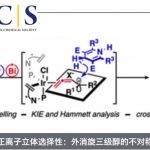
反应机理研究(Fig. 6)。首先,作者对带有苯乙基的噻蒽鎓盐1a、Katritzky盐II以及NHPI酯III进行相关的CV (cyclic voltammogram)研究 (Fig. 6a)。实验观察到,噻蒽鎓盐1a能够显示出不可逆的还原曲线,其中Ered = -1.28 V (versus Ag/AgNO3),因此,与Katritzky盐II(Ered = -1.33 V, versus Ag/AgNO3)以及NHPI酯III (Ered = -1.30 V, versus Ag/AgNO3)相比,1a更易进行相应的还原过程。同时,通过对比1a、B2cat2以及1a与B2cat2混合物的吸收光谱,能够发现相关混合物的吸收光谱出现红移,进而表明反应过程中,能够形成EDA (electron-donor-acceptor)配合物(Fig. 6b)。之后,作者发现,在可见光辐射条件下,采用使用TEMPO或1,1-二苯基乙烯作为自由基捕获剂,能够检测并分离出相应的自由基捕获产物36与37(Fig. 6c)。同时,该小组发现,底物38的脱硫硼化过程,能够形成主要产物39,同时,伴随环化产物40的产生(Fig. 6d)。综上的实验事实表明,反应过程中能够产生相应的烷基自由基中间体。此外,该小组通过“明/暗”实验 (“light/dark”experiment)的研究,进一步表明,可见光为实现上述转化过程的关键因素。接下来,作者通过对产物1c形成过程中,量子产率(Ф = 46)的测定,进一步证实,上述的硼化反应过程中涉及自由基链反应步骤。
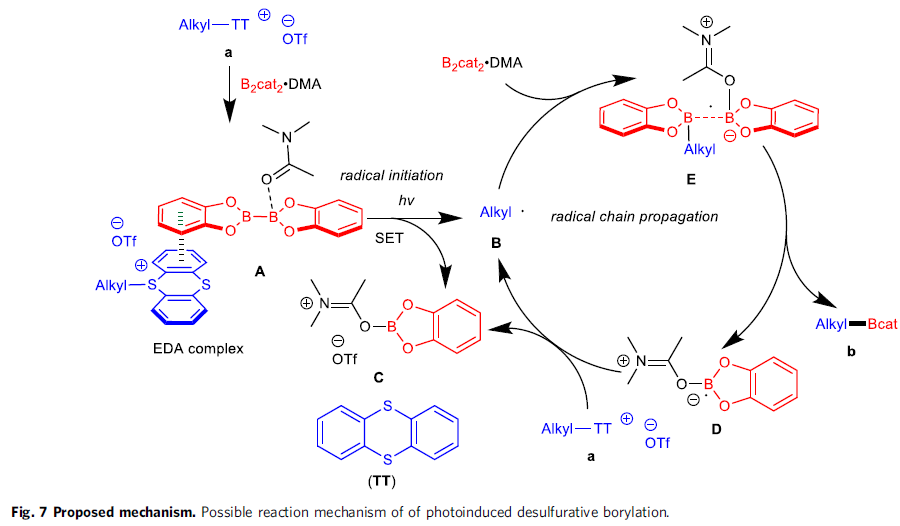
可能的反应机理(Fig. 7)。首先,通过S-(烷基)噻蒽鎓盐与B2cat2·DMA作用,形成EDA配合物A,并通过光激发配合物A的步骤,引发后续的自由基链反应过程,进而形成烷基自由基B、DMA·Bcat加合物C、DMA稳定化的硼中心自由基D以及TT。之后,通过烷基自由基B与B2cat2·DMA加合物之间作用,形成自由基配合物E,继而完成相关的链传递过程。接下来,通过B-B键的断裂,进一步产生硼化产物b与硼基自由基D,并通过硼基自由基D对底物a的进一步还原,形成DMA·Bcat加合物C,同时,使烷基自由基B再生。
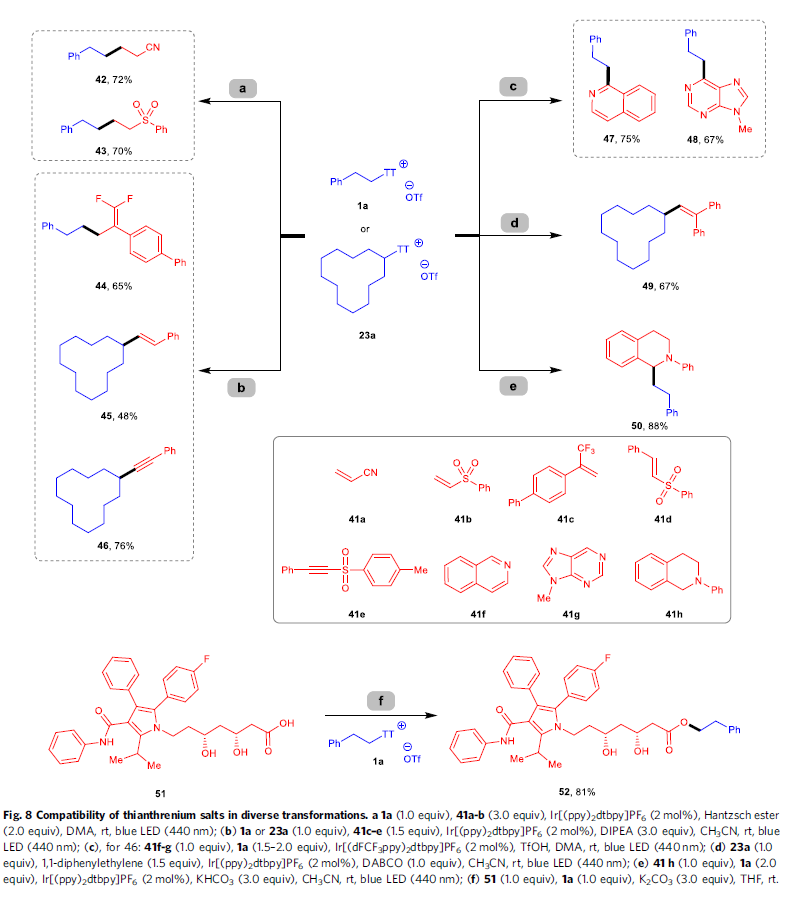
除上述无金属条件下进行的去硫硼化反应方法学之外,噻蒽鎓盐同样能够在室温以及光氧化还原催化的条件下进行多种不同类型的偶联反应过程(Fig. 8)。例如,采用Ir[(ppy)2dtbpy]PF6作为光催化剂,Hantzsch酯作为还原剂,在蓝光LED辐射条件下,噻蒽鎓盐1a能够与缺电子烯基化合物41a与41b经历Giese自由基加成过程,并获得产物42–43。同样地,在采用相同光催化剂Ir[(ppy)2dtbpy]PF6的条件下,通过噻蒽鎓盐1a作为自由基前体,继而与α-三氟甲基烯基化合物41c进行有效的去氟化烷基化 (defluorinative alkylation)反应过程,并获得偕二氟代烯基化合物 (gem-difluoroalkene)44。并且,在相同反应条件下,二级噻蒽鎓盐23a同样能够与砜试剂41d与41e进行后续的烷基自由基烯基化以及炔基化反应,并分别获得目标产物45与46。接下来,作者发现,采用Ir[dF(CF3)ppy]2(dtbpy)PF6作为光催化剂时,并在蓝光LED辐射的条件下,异喹啉(41f)以及9-甲基-9H-嘌呤(41g)均能够有效地与1a进行相应的光氧化还原Minisci型官能团化反应,并分别获得烷基化产物47–48。并且,在可见光辐射条件下,噻蒽鎓盐底物23a能够与1,1-二苯基乙烯进一步反应,并获得相应自由基捕获产物49。此外,N-芳基四氢异喹啉中的C(sp3)-H键同样能够进一步与噻蒽鎓盐1a在可见光光氧化还原体系中,通过直接的偶联过程,以优良的反应收率获得相应产物50。同时,S-(烷基)噻蒽鎓盐能够进一步作为亲电试剂,与亲核试剂进行一系列不同类型的合成转化过程。例如,选择噻蒽鎓盐1a与具有多重反应活性位点的复杂分子atorvastatin(51) 进行反应时,最终能够以优良的选择性与良好的反应收率,获得相应的酯化产物52。
总结
通过S-(烷基)噻蒽鎓盐的选择,能够有效地解决采用锍盐形成不稳定烷基自由基的合成设计方案中存在的巨大挑战。同时,各类具有不同官能团取代的噻蒽鎓盐底物作为有效的亲电试剂,能够成功地应用于光催化条件下的硼化、杂芳基化、烷基化、烯基化以及炔基化反应策略的设计。此外,通过上述策略,同样能够顺利实现羟基向其它类型官能团的转化,尤其一锅反应策略的成功设计,能够近乎完全地回收相应的TT试剂。












No comments yet.