

本文作者:杉杉
导读
近日,慕尼黑工业大学(Technische Universität München)Thorsten Bach课题组在Angew. Chem. Int. Ed.上发表论文,使用10 mol%的手性光敏剂(sensitizer),于420 nm波长照射下,实现3-取代的喹喔啉酮(Quinoxalinones)与苯乙烯的不对称Aza Paternò-Büchi反应,获得14个产物,产率为50-99%,ee为86-98%。同时,作者认为该反应的机理是亚胺底物和催化剂之间发生了三重态能量转移。
Enantioselective, Visible Light-mediated Aza Paternò-BüchiReactions of Quinoxalinones
Xinyao Li, Johannes Großkopf, Christian Jandl, and Thorsten Bach
Angew. Chem. Int. Ed. ASAP DOI:10.1002/anie.202013276 https://doi.org/10.1002/ange.202013276

正文
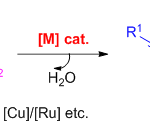
四元环的产生,是光化学标志性的转化之一。目前,烯酮的[2+2]光环加成反应(photocycloaddition)已经被用作合成环丁烷的工具,Paternò-Büchi反应被用作合成氧杂环丁烷的工具(Scheme 1)。然而,对于通过[2+2]光环加成反应合成氮杂环丁烷的研究相对较少。而光化学合成氮杂环丁烷的主要挑战是寻找合适的生色团(chromophores),该生色团易被促进成反应性的激发态,从而形成C-N和C-C键。若将亚胺本身用作生色团,则其取代模式应避免在激发态下发生氧化和避免C-N键的旋转(E/Z异构化)。因此,关于光化学合成氮杂环丁烷的早期工作[1]是环状亚胺的使用,其中许多环状亚胺在氮原子上带有吸电子基团(如rac-1)。最近,Maruoka[2]课题组研究表明,N-磺酰基亚胺也为合适的底物,可能通过单线态激基复合物(singlet exciplex)与苯乙烯发生Aza Paternò-Büchi反应(rac-II)。此外,在Aza Paternò-Büchi反应中,烯烃可作为生色团。Sivaguru[3]课题组以呫吨酮(xanthone)作为三重光敏剂于 350 nm的条件下进行辐照,可得到产物rac-III。Schindler[4]课题组使用铱催化剂作为作光敏剂(427 nm),通过芳基烯烃合成产物rac-IV,这些芳基烯烃通过三原子束缚剂与肟(和腙类)连接。近期,Schindler[5]课题组提出了使用2-异恶唑啉-3-羧酸盐作为亚胺生色团和铱配合物作为光敏剂,可能实现分子间的Aza Paternò-Büchi反应。

迄今为止,大多数光化学合成的氮杂环丁烷均为外消旋混合物,基于轴向手性酰胺的使用是获得对映体富集化合物有效方法,该手性可在Aza Paternò-Büchi反应过程中进行手性转移。尽管有阻转选择性(atropselective)的光化学反应,但它们依赖于手性底物的使用,不能提供催化对映选择性过程的选择。在此,慕尼黑工业大学Thorsten Bach课题组报道了一种通过使用手性三重态光敏剂,实现喹喔啉酮与苯乙烯的不对称Aza Paternò-Büchi反应,从而合成光学纯的手性氮杂环丁烷化合物。
首先,作者以3-甲基-喹喔啉酮(1a)作为亚胺组分,苯乙烯为烯烃,进行了相关Aza Paternò-Büchi反应的条件筛选(Table 1)。反应的最佳条件为:使用10 mol%的光敏剂2,于420 nm波长辐射下,在-25 ℃的DCE中反应,即可获得98%收率和93% ee的目标产物3a。

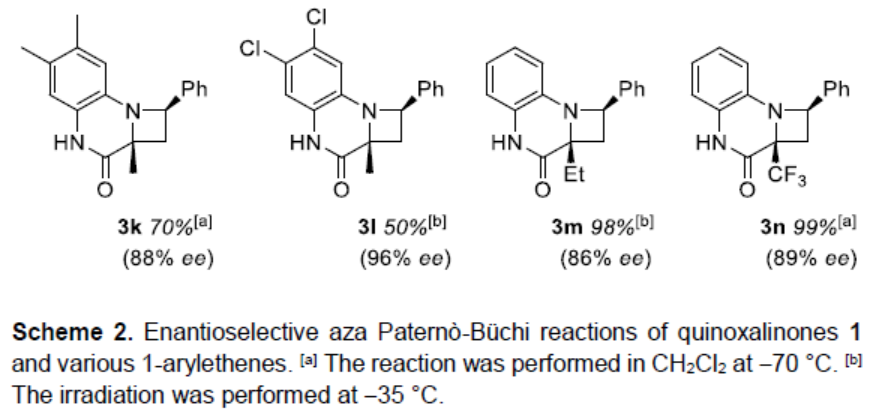
在获得上述最佳反应条件后,作者开始对喹喔啉酮和烯烃底物进行了扩展(Scheme 2)。苯乙烯的芳基取代不受电子效应和定位效应的影响,含有Me、tBu、卤素时,均以优异的收率(90-99%)和高对映选择性(91-98%ee)获得产物3b-3i。同时,含有4-吡啶基的烯烃同样取得良好的结果(3j)。此外,由于具有不同取代的喹喔啉酮不易合成,所以选择了四个底物进行研究,均可与体系兼容,从而获得产物3k-3n。
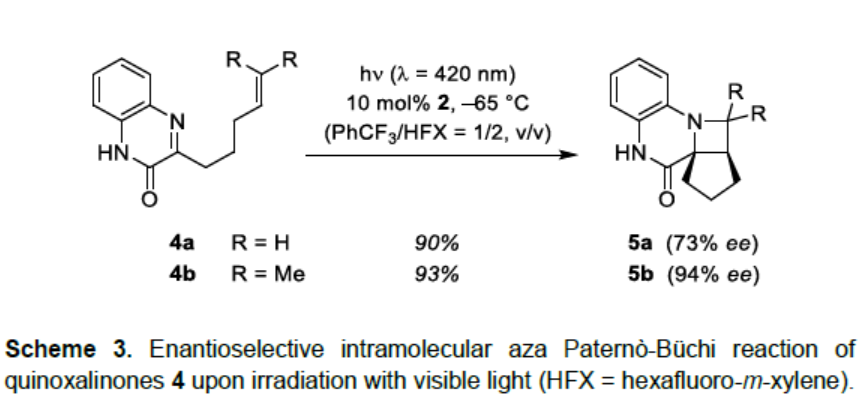
紧接着,作者对分子内的Aza Paternò-Büchi反应进行了研究(Scheme 3)。以化合物4为底物,使用非手性9-噻吨酮(TXT)作为光敏剂,可生成预期的四环产物rac-5。若使用手性噻吨酮2时,则以相同的收率和高对映选择性获得产物5。在这种情况下,底物在非极性溶剂中的溶解度高于与喹喔啉酮1进行分子间反应。因此,使用三氟甲苯和六氟间二甲苯(HFX)的混合溶剂,可在低至-65 ℃的温度下进行辐照。在这些条件下,产物5b的对映选择性很高(94%ee)。而电子富集较少的末端烯烃4a,获得的产物5a的对映选择性偏低(73%ee)。
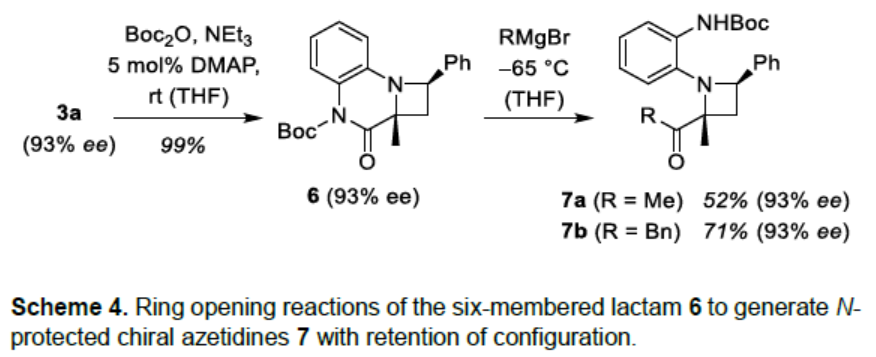
随后,作者对产物3a进行了衍生化(Scheme 4)。首先,将内酰胺3a进行Boc保护,形成化合物6,再与格氏试剂进行亲核进攻,从而获得N-保护的氮杂环丁烷7。值得注意的是,所有转化均未对光学纯度造成影响。
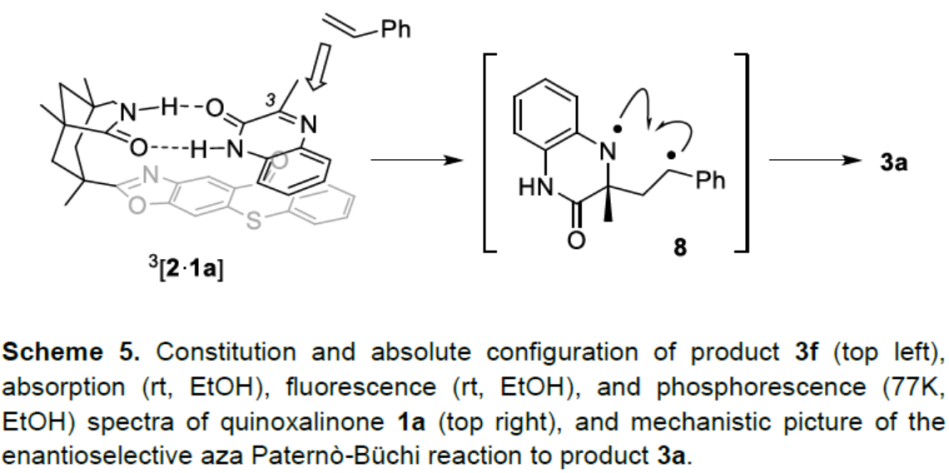
为了进一步确认氮杂环丁烷3的相对构型,作者进行了单晶培养,从而获得了化合物3f的晶体(Scheme 5)。如图所示,氮杂环丁烷中的芳基环和甲基取代基为顺式,因为反式构型会引起两个芳环的空间排斥。而异常衍射数据能够评估产物的绝对构型,如喹喔啉酮的C3位置的C-C键形成在其Re面上。与先前对催化剂2的研究相一致,氢易于与底物1a结合,从而促进能量转移以及配合物3[2•1a]中C-C键的形成。1a的Si面被噻吨酮实体所屏蔽,后者只能让苯乙烯仅攻击Re面。中间的1,4-双自由基8在非对映选择性(ISC)后形成氮杂环丁烷产物3a。尽管从三重态2到苯乙烯的能量转移在能量上是可行的,但这种途径不太可能发生,很难解释为什么对映选择性如此之高,而催化剂的负载量仅为10 mol%。另外,应优选2与喹喔啉酮的配合物的分子内能量转移。通过热力学从喹喔啉酮单电子转移的替代反应途径是不利的。尽管喹喔啉酮的三重态可以被还原剂淬灭,但苯乙烯还原是不可行的。此外,通过化合物1a的基态还原电位,从而表明三重态仅是一种弱氧化剂。
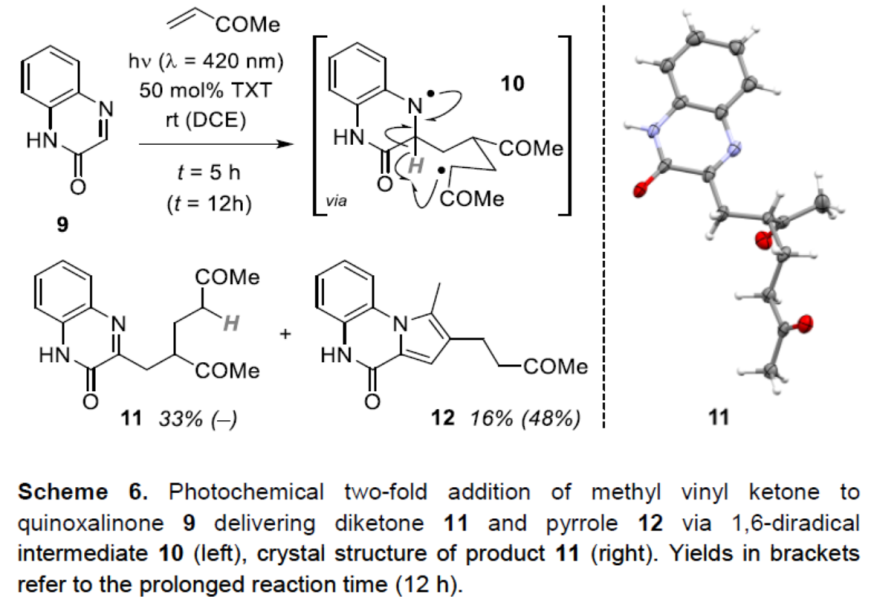
此外,与3-烷基化的底物1和4不同,在C3位置未取代的喹喔啉酮9与苯乙烯进行反应时,未生成氮杂环丁烷产物,而形成了C3取代产物(Scheme 6)。而使用甲基乙烯基酮时,以TXT为非手性光敏剂进行反应,得到两种产物的混合物。反应5小时后,以33%的收率获得产物11。若继续反应,则分离出另一种产物,可能为吡咯12(可能由Paal-Knorr型缩合反应形成)。产物11的形成表明,与8相连的1,4-二自由基并不接近氮杂环丁烷环,而是在C-末端加成到甲基乙烯基酮的第二个分子上。所得的1,6-二自由基10在C3碳原子进行γ-抽氢反应。不幸的是,11的稳定性有限,导致未能进行其他不对称反应的验证。
总结
慕尼黑工业大学Thorsten Bach课题组报道了使用10 mol%的手性光敏剂,于420 nm波长照射下实现3-取代的喹喔啉酮(亚胺)与苯乙烯(烯烃)的不对称Aza Paternò-Büchi反应。其中,喹喔啉酮的内酰胺部分能够与催化剂直接进行氢键的相互作用,导致三重态能量转移和有效的对映体分化。而芳基烯烃用作烯烃底物,不仅保证立体异构中心的快速形成,而且具有高的非对映选择性。
参考文献
[1] Selected examples: a) O. Tsuge, M. Tashiro, K. Oe, Tetrahedron Lett. 1968, 9, 3971-3974; b) J. A. Hyatt, J. S. Swenton, J. Chem. Soc., Chem. Commun.1972, 1144-1145; c) T. H. Koch, K. H. Howard, Tetrahedron Lett.1972, 11, 4035-4038; d) O. Tsuge, K. Oe, M. Tashiro, Tetrahedron 1973, 29, 41-46; e) J. S. Swenton, J. A. Hyatt, J. Am. Chem. Soc. 1974, 96, 4879-4885; f) K. A. Howard, T. H. Koch, J. Am. Chem. Soc. 1975, 97, 7288-7298; g) R. M. Rodehorst, T. H. Koch, J. Am. Chem. Soc. 1975, 97 7298-7304; h) S. Futamura, H. Ohta, Y. Kamiya, Bull. Chem. Soc. Jpn. 1982, 55, 2190-2194; i) T. Nishio, J. Org. Chem. 1984, 49, 827-832; j) T. Nishio, J. Chem. Soc., Perkin Trans. 1, 1990, 565-570.[2] R. Sakamoto, T. Inada, S. Sakurai, K. Maruoka, Org. Lett. 2016, 18, 6252-6255.
[3] E. Kumarasamy, S. K. Kandappa, R. Raghunathan, S. Jockusch, J. Sivaguru, Angew. Chem. 2017, 129, 7162-7167.; Angew. Chem. Int. Ed. 2017, 56, 7056-7061.
[4] M. R. Becker, A. D. Richardson, C. S. Schindler, Nat. Comm. 2019, 10, 5095-5102.
[5] M. R. Becker, E. R. Wearing, C. S. Schindler, Nat. Chem. 2020, 12, in press, DOI: 10.1038/s41557-020-0541-1.
















No comments yet.