
本文作者:杉杉
导读
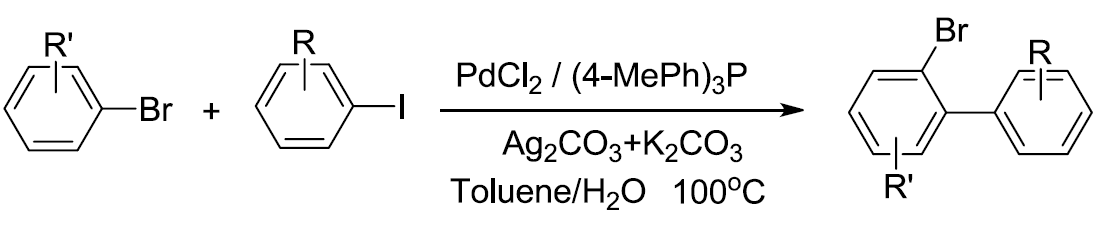
芳基溴是有机化学中最为重要的化合物之一,通过C-Br键能够实现多种类型的合成转化,进而完成一系列具有重要应用价值的复杂有机分子的构建。其中,芳基溴的直接C-H键官能团化方法学是构建官能团化芳基溴分子的有效方法,然而,由于芳基溴参与C-H键活化时,反应性相对较低,因此,采用芳基溴底物参与的C-H键活化过程,较少有相关的文献报道。近日,南京工业大学张宏海课题组在Organic Letters中发表论文,报道采用钯催化的芳基碘与芳基溴之间的交叉偶联反应方法学,进而完成一系列溴代联芳基化合物的合成。其中,反应机理涉及银媒介促进的C-H键活化步骤。
C-H Bond Functionalization of (Hetero)aryl Bromide Enabled Synthesis of Brominated Biaryl Compounds
K. Liu, G. Hu, C. Wang, F. Sheng, J. Bai, J. Gu, H Zhang, Org. Lett. ASAP. doi: 10.1021/acs.orglett.1c01613.
正文
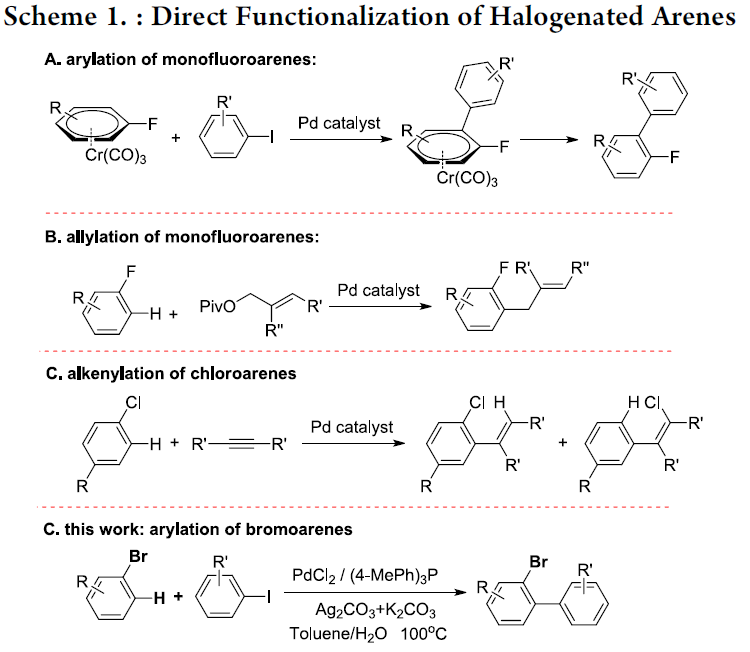
近年来,过渡金属催的化C-H键官能团化方法学能够高效地完成一系列不同结构的有机分子的构建,因而备受关注。在扩展C-H键官能团化方法学底物应用范围的研究中,多数情况下,需要采用导向基团的螯合辅助,进而提高反应活性与选择性。然而,对于缺乏导向基团辅助的简单芳烃的直接C-H键官能团化方法学,文献报道则较为有限[1]。Fagnou课题组[2]报道首例通过高度缺电子多氟芳烃参与的直接C-H芳基化过程,其反应机理涉及协同金属化去质子化 (concerted metalation deprotonation,CMD)过程。之后,研究发现,通过CMD机理同样能够成功完成各类五元杂芳烃、二嗪N-氧化物以及硝基芳烃衍生物的直接C-H芳基化反应。另一方面,对于具有单个或两个卤原子取代的芳烃底物而言,要实现直接的C-H芳基化过程,则面临巨大的挑战。最近,Larrosa课题组[3]报道钯催化条件下,通过π-配位Cr(CO)3单元辅助的单氟芳烃的直接芳基化反应方法学 (Scheme 1)。2016年,Hartwig课题组[4]报道了通过钯催化剂以及AgOPiv参与的单氟芳烃的直接烯丙基化反应方法学。此外,Duan课题组[5]在钯催化条件下,顺利实现芳基氯底物的C-H烯基化反应方法学。然而,对于通过C-H键活化途径,实现芳基溴直接官能团化的研究,则较少有文献报道。最近,本课题组[6] 研究发现,在碳酸银催化剂存在下,芳基溴能够进行高效的H/D交换反应,进而表明采用银盐能够有效地促进芳基溴的直接C-H键活化过程。这里,本文将报道一种通过钯催化剂促进的,芳基溴与芳基碘之间的交叉偶联反应方法学,进而完成一系列溴代联芳基化合物合成。
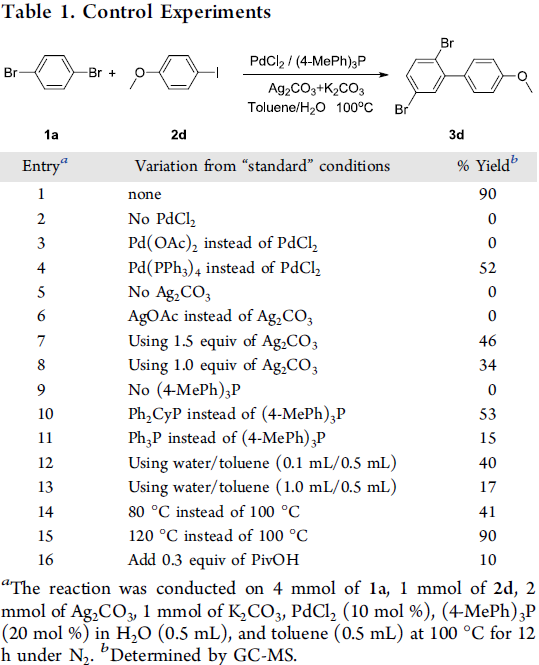
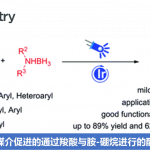
首先,作者采用1,4-二溴苯1a与4-碘苯甲醚2d作为模型底物,进行了相关偶联反应条件的优化筛选 (Table 1)。确定最佳的反应条件为:采用PdCl2作为催化剂,(4-MeOh)3P作为膦配体,Ag2CO3作为银盐,K2CO3作为碱,在甲苯与水的混合溶剂中,100℃下进行反应,最终获得90%收率的偶联产物3d。
在上述最佳反应条件下,作者首先对芳基碘的底物应用范围进行考察 (Scheme 2)。研究表明,一系列带有供电子基与吸电子基取代的芳基碘底物均能较好地与上述反应条件兼容,并获得相应的产物3a–3n,收率为46-97%。值得注意的是,由于卤素取代以及立体效应的影响,3e、3f以及3n收率出现显著降低。同时,研究发现,稠合的芳基碘底物同样能够有效地参与上述的C−H 键芳基化过程,并以55-75%收率,获得相应产物3o–3q。此外,3-碘吡啶同样能够较好地与上述反应体系兼容,并获得57%收率的偶联产物3r。
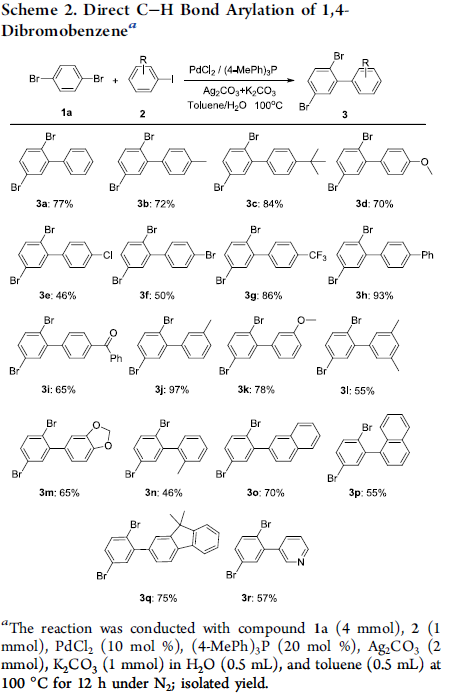
接下来,作者对芳基溴底物的应用范围进行考察 (Scheme 3)。研究表明,1,4-二溴萘、各类取代的1,4-二溴苯以及1,3,5-三溴苯均能够与1-(叔丁基)-4-碘苯顺利地进行相应的C-H芳基化过程,并获得相应的偶联产物4a–4d,收率为60-98%。然而,作者发现,在采用1-溴-4-(三氟甲基)苯作为底物时,却获得两种偶联产物的异构体,收率为55%,产物比为19:1。同时,作者发现,上述反应条件对于其它单溴代芳烃,则无法良好地兼容,并观察到产物收率或异构体比率较低。此外,该小组进一步发现,溴代吡啶衍生物同样能够良好地应用于上述的C-H芳基化过程,并以优良的反应收率获得相应的偶联产物4f–4j。
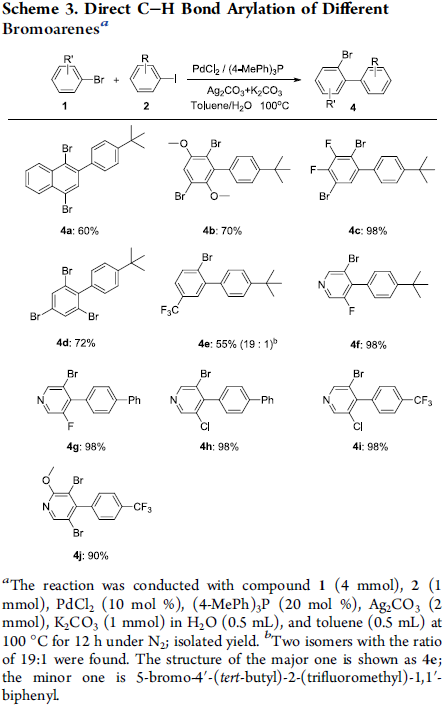
为了进一步研究上述C-H芳基化反应的实用性,作者进一步对获得的芳基化产物进行相应的后期修饰 (Scheme 4)。首先,作者发现,将底物3-溴-5-氟吡啶的用量扩大至10 mmol,同样能够获得98%收率的芳基化产物4f。同时,作者进一步观察到,芳基化产物4f中的C-Br键能够进一步经历一系列不同的偶联反应过程,并获得化合物5a–5c。
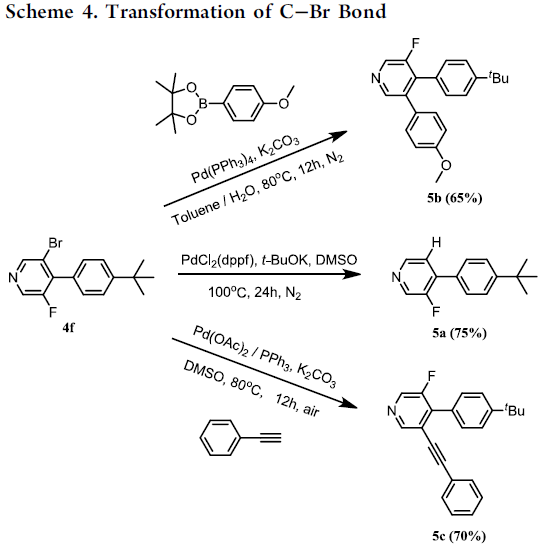
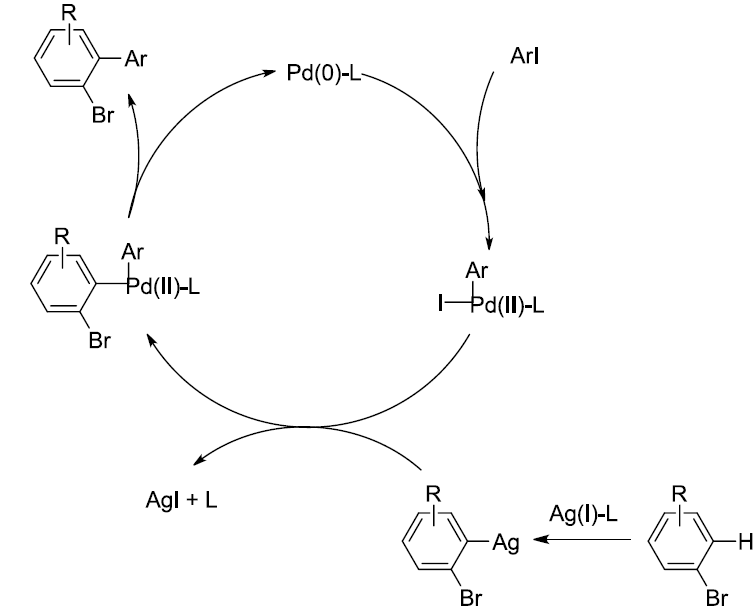
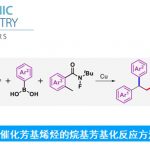
为了提出合理的反应机理,作者进行一系列相关的平行实验 (parallel experiment)研究 (Scheme 5)。首先,在无PdCl2存在的条件下,采用D2O与甲苯的混合溶剂进行相关反应时,能够获得84%氘掺入 (deuterium incorporation)的1,4-二溴苯产物。相反地,作者发现,反应体系中无Ag2CO3存在时,仅能够回收未氘代的起始原料。上述实验结果表明,碳酸银可能参与C-H键的活化步骤。接下来,在动力学同位素效应 (KIE) 的研究中,作者观察到kH/kD为3.3,表明决速步骤中可能涉及C-H键的断裂。基于上述研究以及前期的文献报道,作者提出了一种合理的反应机理路径。首先,芳基碘与Pd(0)配合物进行氧化加成过程,形成芳基钯(II)中间体。同时,芳基溴底物通过Ag配合物通过C-H键的活化过程,形成芳基银(I)中间体。接下来,芳基钯(II)中间体与芳基银(I)中间体通过碱促进的转金属化过程,形成二芳基钯配合物。最终,通过二芳基钯配合物的还原消除过程,获得相应的溴代联芳产物,并使Pd(0)催化剂再生。
总结
南京工业大学张宏海课题组成功开发出通过PdCl2/(4-MePh)3P催化体系促进的芳基溴直接C-H键芳基化反应方法学策略,进而顺利完成一系列溴代联芳基化合物的构建。同时,通过平行实验研究表明,碳酸银的加入,对于C-H键的活化步骤的顺利进行尤为关键。此外,作者进一步研究发现,芳基化产物中的C-Br键能够通过后续的反应过程,进一步转化为具有多种不同官能团的重要合成砌块。并能够将上述砌块应用于一系列复杂药物分子与功能材料的合成。
参考文献
[1] (a) P. Wedi, M. van Gemmeren, Angew. Chem. Int. Ed. 2018, 57, 13016. doi: 10.1002/anie.201804727.(b) T. W. Lyons, M. S. Sanford, Chem. Rev. 2010, 110, 1147. doi: 10.1021/cr900184e 10.1021/cr900184e.
[2] M. Lafrance, C. N. Rowley, T. K. Woo, K. Fagnou, J. Am. Chem. Soc. 2006, 128, 8754. doi: 10.1021/ja062509l. [3] (a) P. Ricci, K. Kramer, X. C. Cambeiro, I. Larrosa, J. Am. Chem. Soc. 2013, 135, 13258.doi:10.1021/ja405936s.(b) D. Whitaker, J. Bures, I. Larrosa, J. Am. Chem. Soc. 2016, 138, 8384. doi: 10.1021/jacs.6b04726.
[4] S. Y. Lee, Hartwig, J. F. J. Am. Chem. Soc. 2016, 138, 15278. doi: 10.1021/jacs.6b10220. [5] Z. Li, W. Duan, Angew. Chem. Int. Ed. 2018, 57, 16041. doi: 10.1002/anie.201808866. [6] G. Hu, J. Bai, E. Li, K. Liu, F. Sheng, H. Zhang, Org. Lett. 2021, 23, 1554. doi: 10.1021/acs.orglett.0c04139.


















No comments yet.