
本文作者:ChemBoy
导读
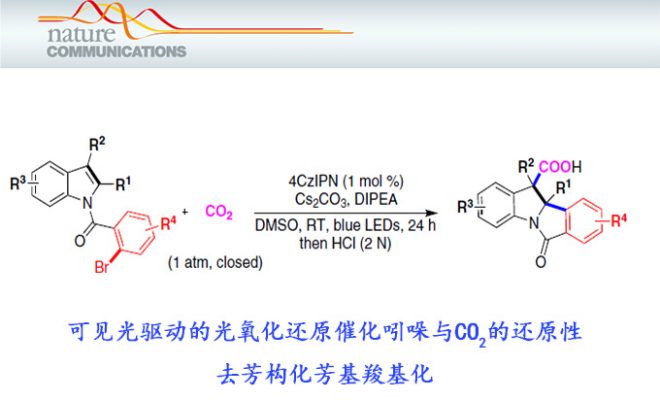
最近,四川大学余达刚教授课题组在Nat. Commun.中报道了通过可见光诱导光氧化还原催化的、以CO2为羧基源,进行的吲哚类底物C2=C3双键的去芳构化芳基羧基化双官能团化反应。该报道是选择性的串联还原环化/交叉偶联反应方法学的典型代表[1],为去芳构化的双官能团化反应研究开辟了新的途径。
“Reductive dearomative arylcarboxylation of indoleswith CO2 via visible-light photoredox catalysis”
Wen-Jun Zhou, Zhe-Hao Wang, Li-Li Liao, Yuan-Xu Jiang, Ke-Gong Cao, Tao Ju, Yiwen Li,
Guang-Mei Cao & Da-Gang Yu
Nat. Commun. 2020, 11, 3263.DOI:10.1038/s41467-020-17085-9
正文
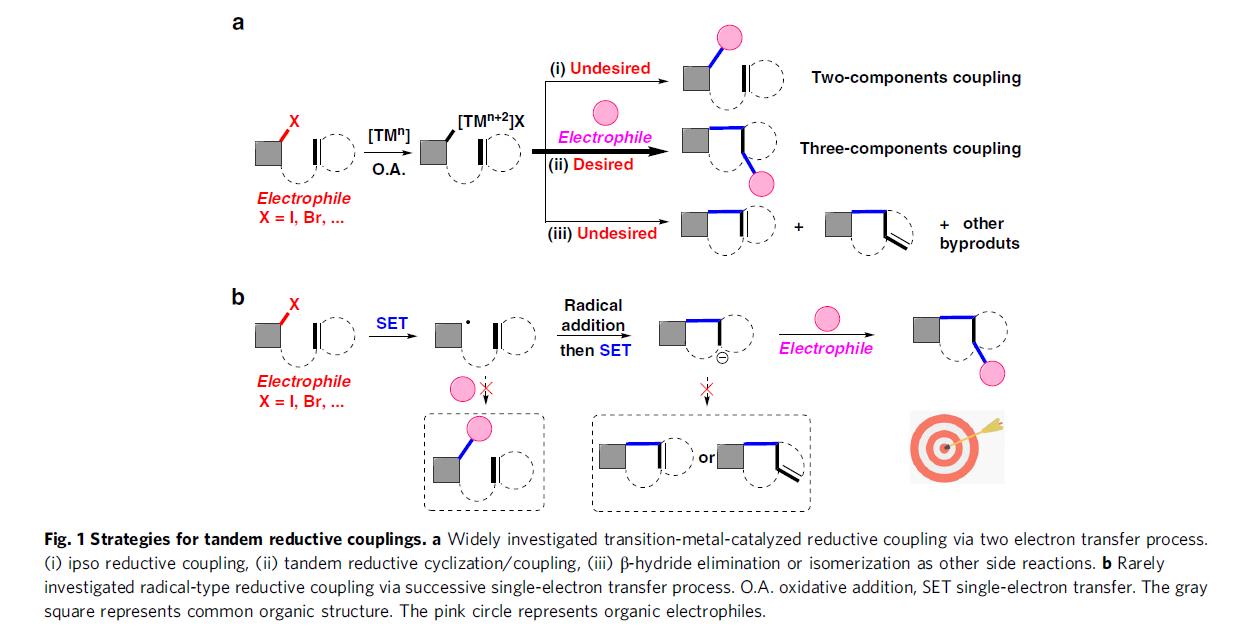
交叉亲电偶联反应已经成为构筑C-C键的一种有力方法[2]。与传统过渡金属催化的还原偶联相比,交叉亲电偶联具有底物易得、反应操作简单与步骤经济的优势 (Fig. 1a, path i)[3]。 近年来,对于三组分的还原性偶联反应的研究已取得重大进展。通过不饱和键与两种亲电试剂之间的双官能团化反应 ,能够同时形成两种新的化学键,进而迅速构建起高度官能团化的分子骨架 (Fig. 1a, path ii)[4]。值得注意的是,如果其中一种亲电试剂能够与不饱和键实现连接,便可以通过分子内环化反应构筑新的环系[5]。在串联的还原环化交叉偶联反应中,尽管通过2e-转移过程进行的过渡金属催化,可以很好的调节反应活性与选择性。然而,将其应用于串联的还原环化交叉偶联反应中仍存在一些挑战。例如,如果不饱和键的反应活性不高,迁移插入步骤不够快,则有利于亲电试剂的本位官能团化 (Fig. 1a, path i);此外,反应过程中产生的金属有机中间体还能够进一步发生质子化、β-H消除或异构化等副反应(Fig. 1a, path iii)[1]。因此,有必要发展一种能够抑制上述副反应,且具有高度选择性的新策略,例如连续单电子转移(SSET)策略(Fig. 1b)。
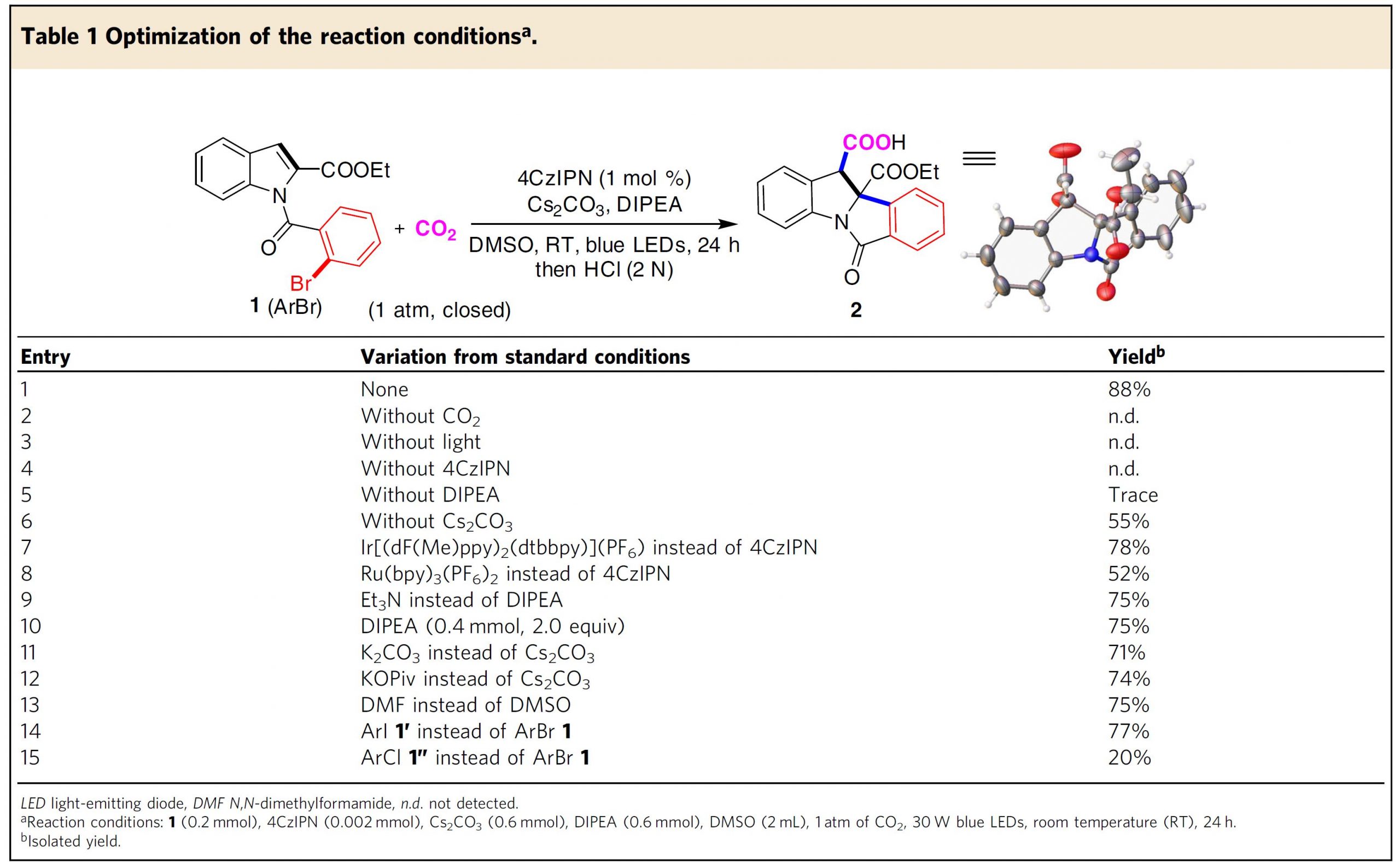
基于以上设计与假设,作者首先以1-(2-溴苯甲酰基)-1H-吲哚-2-羧酸乙酯1作为模板底物,在CO2 (1 atm)气氛及可见光辐射条件下,对反应条件进行筛选优化 (Table 1)。最终,作者确定最佳的反应条件为:以4CzIPN (1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene)作为光催化剂、Cs2CO3作为碱、DIPEA作为电子供体以及采用DMSO作为反应溶剂。在上述最佳反应条件下,模板底物能够以88%的分离产率与较高的非对映选择性 (d.r. >19:1).获得相应吲哚羧酸产物2 (Table 1, entry 1)。同时,作者通过控制实验表明CO2、可见光、光催化剂与还原剂对该合成转化的顺利进行,均较为关键 (Table 1, entries 2-5);作者同样对其它类型的光催化剂如Ir-和Ru-配合物进行了进一步研究,最终发现采用这类催化剂时,目标产物产率偏低(Table 1, entries 7 and 8);用Et3N代替DIPEA作为还原剂或将DIPEA由3.0eq.减少至2.0 eq.时,观察到产率降低至75% (Table 1, entries 9 and 10);此外,研究发现,采用其它碱, 如K2CO3与KOPiv时,该反应仍然可以有效地进行,然而,目标产物产率有所降低 (Table 1, Entries 11 and 12);同时,作者发现改用DMF作为反应溶剂时,同样能够顺利获得预期的目标产物,然而,与DMSO作为反应溶剂时相比,目标产物产率有所下降 (Table 1, entry 13)。随后,作者进一步对芳基碘与芳基氯底物进行深入研究,发现与芳基溴相比,目标产物产率显著降低 (Table 1, entries 14 and 15)。并且,反应过程中未观察到芳基卤化物的本位羧基化产物。

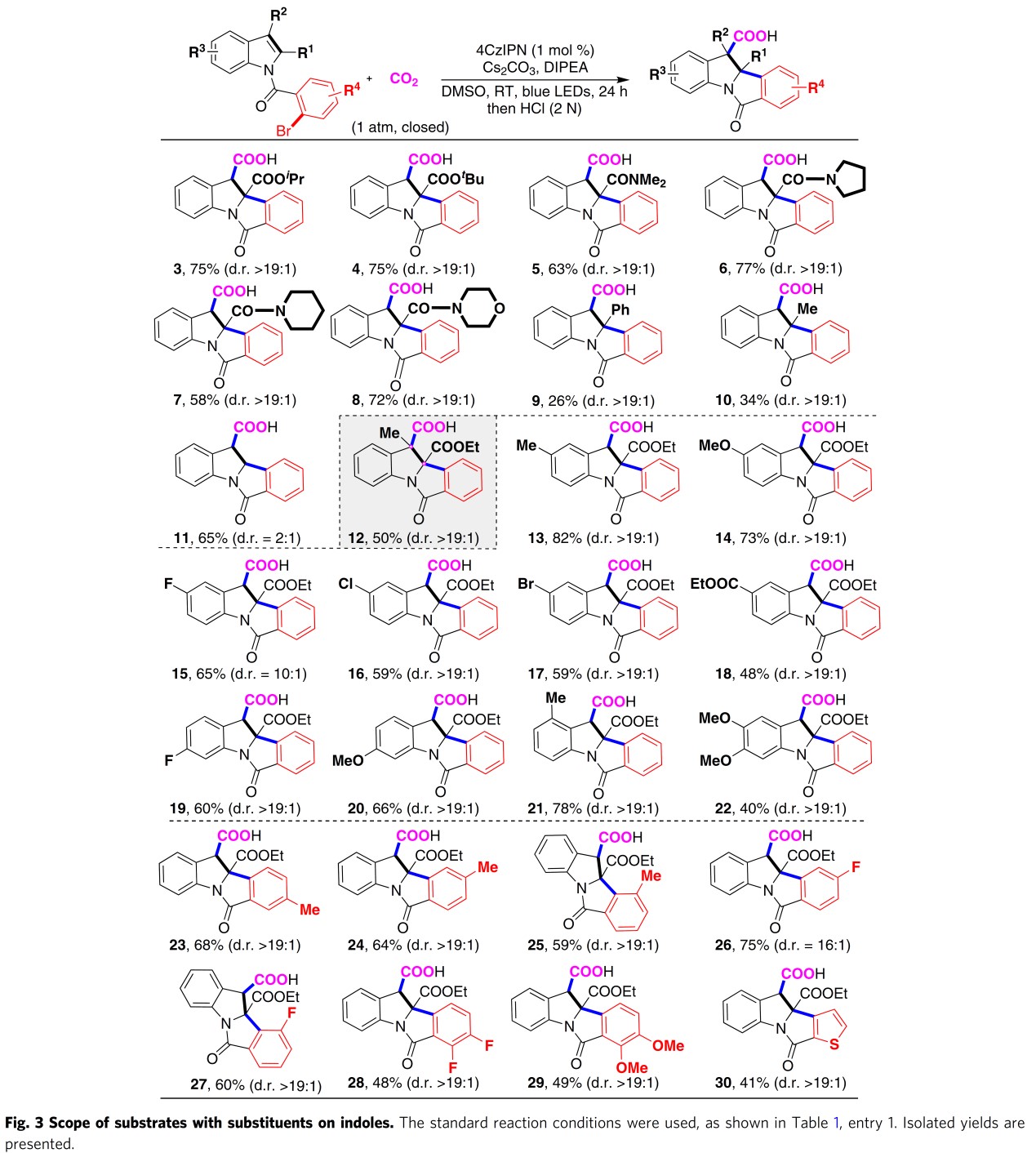
接下来,在最佳反应条件下,作者首先对吲哚环中取代基的电子效应进行了系统研究(Fig. 3),发现在大多数底物中,反应均具有高度的非对映选择性 (>19:1 d.r)。当吲哚C2位为烷氧羰基取代时,反应活性较高 (2-4);此外,C2位为酰胺基时,同样能够以中等至良好的收率获得相应目标产物 (5-8);然而,当C2位为-Ph (9)与-Me (10)取代时,反应产率明显降低,可能源自芳基自由基与吲哚的自由基加成步骤受阻。当吲哚C2位无取代基时,反应同样能够顺利进行,并以65%的收率获得相应目标产物 (11),然而,却观察到非对映选择性出现显著降低 (11, 2:1 d.r),这表明C2位取代基的存在对于反应过程中非对映选择性的控制尤为关键。值得注意的是,当C2和C3位同时有取代基存在时,能够以中等收率与高度的非对映选择性获得具有双季碳中心的产物 (12)。另外,作者还发现在吲哚环其它位置 (如C5-、C6-)存在供电子取代基或吸电子取代基取代时,均能够以中等至良好的产率以及较高的非对映选择性获得相应目标产物 (13-20)。作者进一步研究表明,上述反应条件对于取代基的立体效应并不敏感。同时,该反应条件对于C4-位存在甲基 (21)以及 C5-以及C6-位二取代的底物 (22), 同样能够较好地兼容。
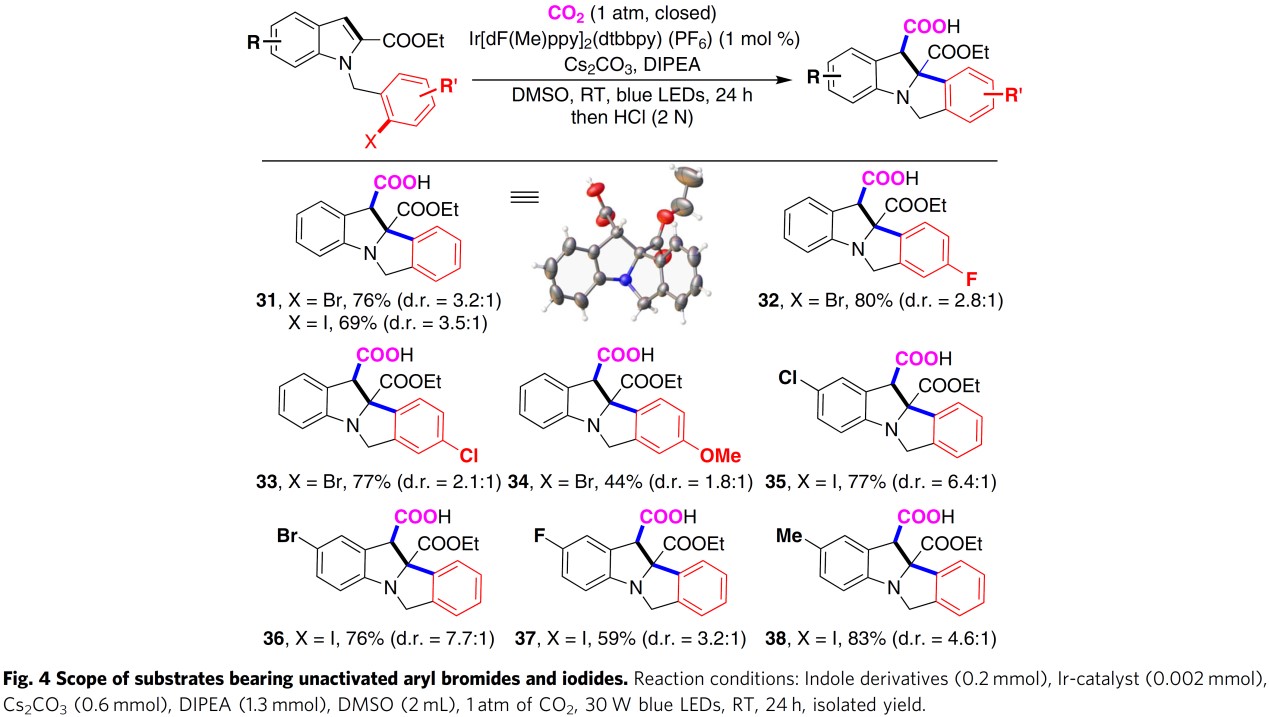
在对吲哚环中不同取代基的影响进行系统研究之后,作者接下来进一步考察芳基溴底物中取代基 (R4)的电子效应。研究表明,芳基溴的对位、间位、邻位存在甲基取代 (23-25),间位与邻位存在氟取代(26-27),二氟取代 (28),以及二甲氧基取代 (29)与杂环稠合 (30)时,均能够以中等至良好的收率、较高的非对映选择性获得相应目标产物。除芳基溴外,作者还对更加富电子的芳卤进行了深入研究 (Fig.4), 进而发现各类取代芳基溴与取代芳基碘均能够以中等至良好的产率获得相应目标产物,然而,仅获得中等程度的非对映选择性 (31-38)。
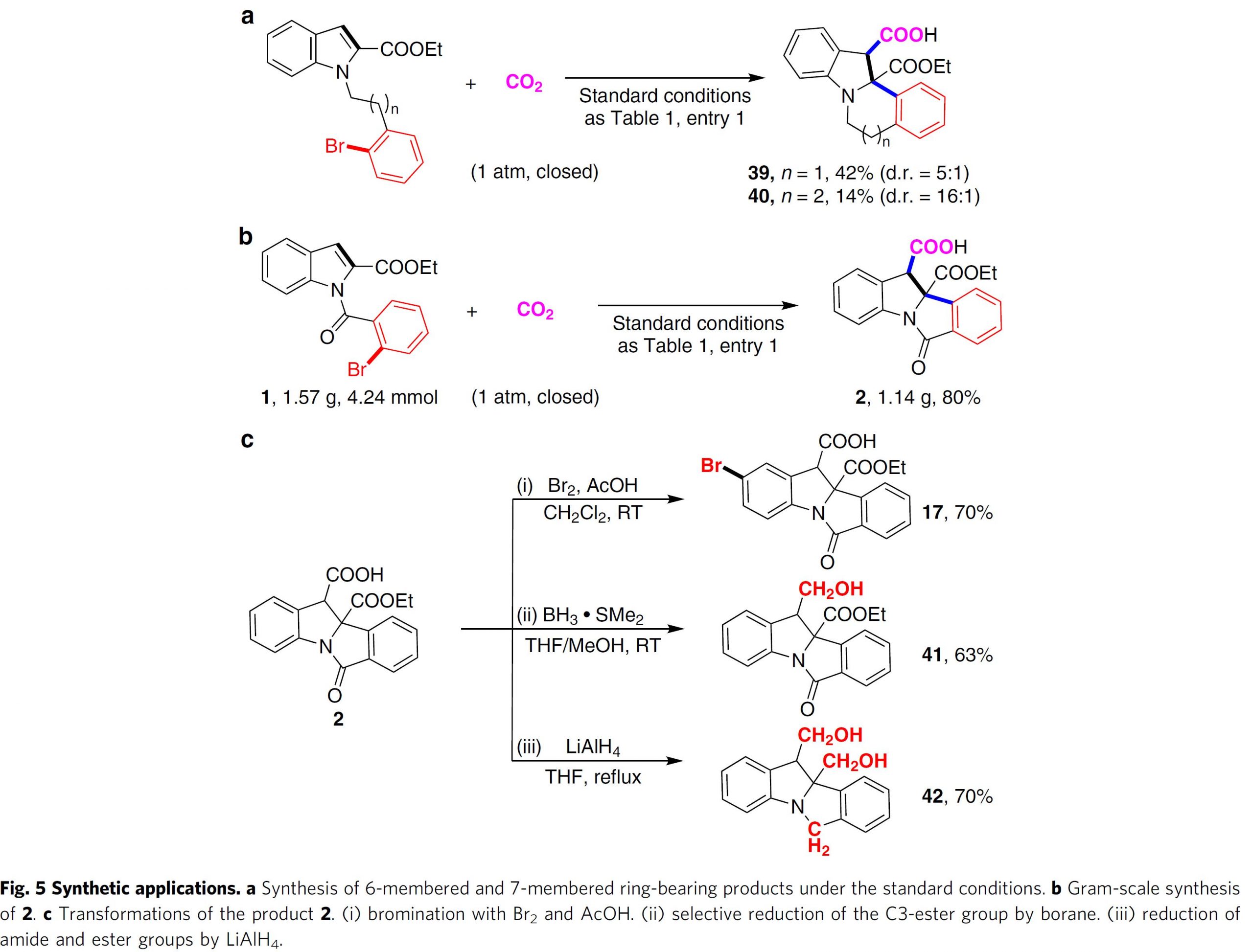
随后,作者对该方法学合成应用进行考察 (Fig. 5)。作者发现,通过延长吲哚环与芳基溴之间的碳链 (Fig. 5a),在标准反应条件下,能够分别以42% (5:1 d.r)、14% (16:1 d.r)的收率与非对映选择性获得相应的六元环 (39)与七元环 (40)产物。之后,作者进一步对模板底物进行克级放大反应研究,观察到在标准反应条件下,能够以80%的收率获得相应目标产物(Fig. 5b)。另外,作者还对产物2进行了后期衍生化研究 (Fig. 5c)。
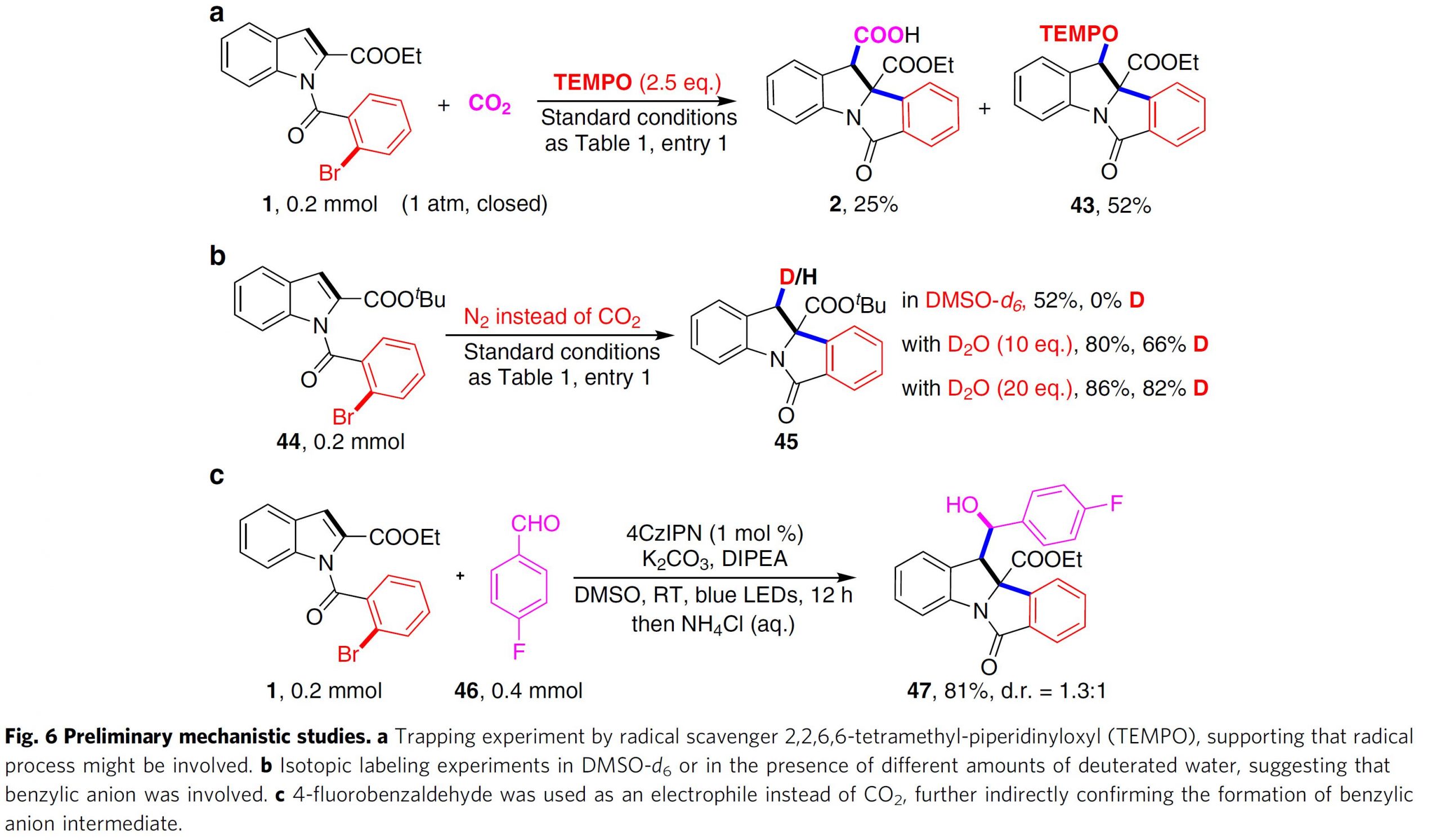
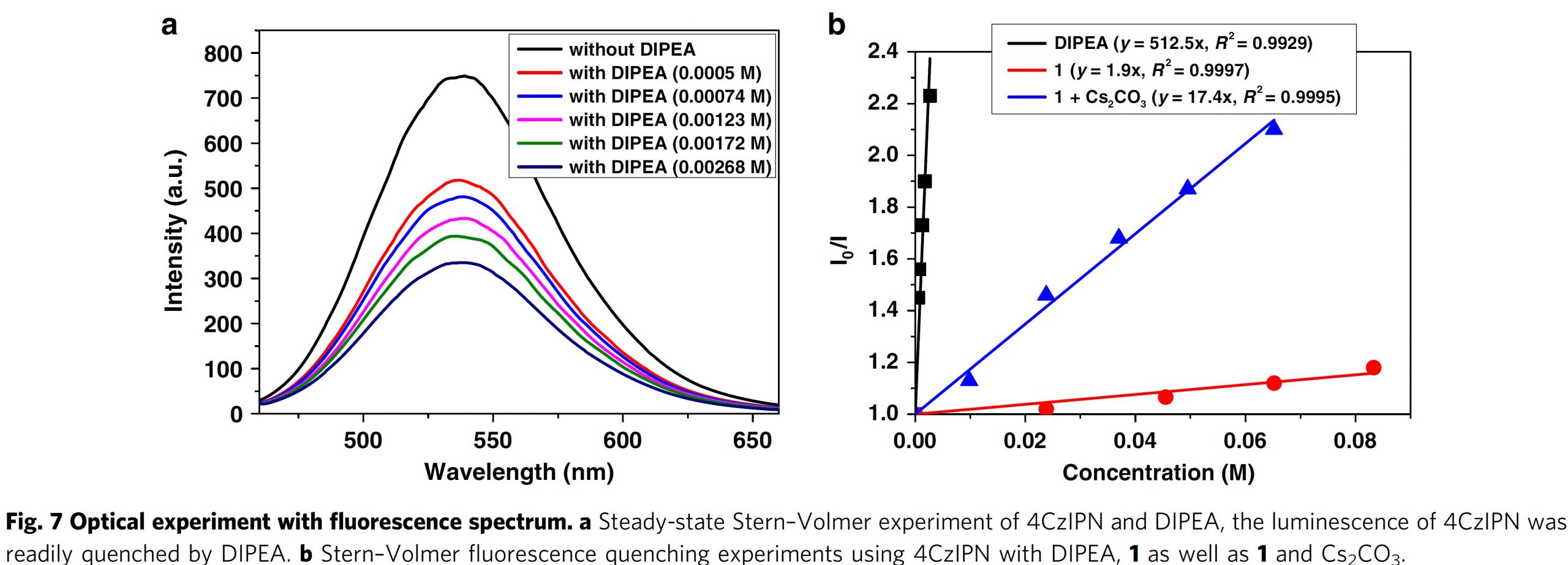
最后,作者对反应机理进行了深入研究。首先,通过自由基捕获实验,证实反应过程涉及苄基自由基的生成 (Fig. 6a);之后,作者在N2气氛以及标准反应条件下进行氘代实验 (Fig. 6b),实验过程中发现,以DMSO-d6作为溶剂进行吲哚化合物44的反应时,未观察到C3位氘代产物45的生成,由此,可以排除通过DMSO进行氢原子转移反应的可能性。而通过加入D2O,在同样条件下进行反应时,却能够观察到有大量氘代产物45出现,进而表明反应过程中涉及苄基碳负离子的参与;而且,在略微改进的反应条件下,采用4-氟苯甲醛代替CO2作为亲电试剂时,发现能够以81%的产率获得化合物47,从而进一步证实反应过程中涉及苄基碳负离子中间体的生成 (Fig. 6c)。此外,作者进一步进行Stern-Volmer荧光猝灭实验,发现4CzIPN在λmax = 536 nm 的荧光能够通过DIPEA (斜率为512.5)的加入而十分容易地发生猝灭。而且,比加入吲哚1 (斜率为1.9)以及吲哚1与碳酸铯 (斜率17.4)的混合物时,猝灭效应更加显著。 综上实验事实,表明DIPEA优先与激发态的4CzIPN 发生SET 过程 (Fig. 7)。
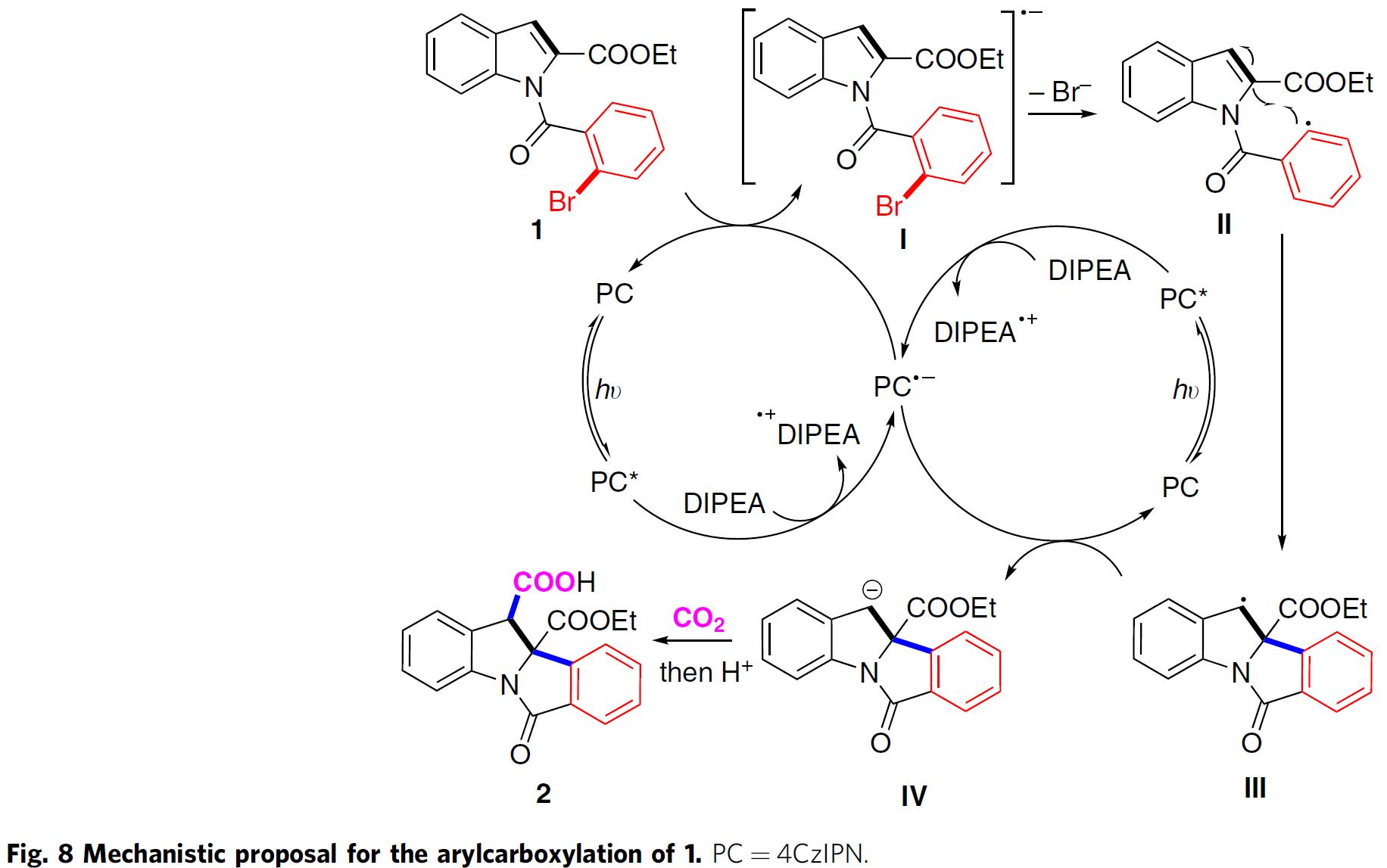
基于上述研究实验与之前的研究报道,作者提出如下可能的反应机理 (Fig. 8)。首先,基态光催化剂4CzIPN (E1/2 [4CzIPN/4CzIPN•−] = −1.21 V vs. SCE in MeCN)在可见光照射下,形成激发态4CzIPN*, 激发态的光催化剂4CzIPN*通过DIPEA (E1/2Ox=+0.63 V vs. SCE in DMF)的加入,进而发生还原猝灭,生成自由基负离子4CzIPN•−与自由基正离子DIPEA•+,底物1与4CzIPN•−发生单电子还原,生成自由基负离子中间体I, 同时,使基态的 4CzIPN催化剂再生,从而完成一次催化循环。接下来,自由基负离子中间体I通过溴离子的离去,形成芳基自由基中间体II,随后,芳基自由基中间体II与吲哚的C2=C3双键发生分子内自由基加成环化,形成苄基自由基中间体III, 苄基自由基中间体III随即与4CzIPN•−发生单电子转移,生成苄基负离子中间体IV, 最后苄基中间体IV与CO2发生亲核加成,再经质子化过程,获得目标产物2。
(注:本文中所有图片均来自Nat. Commun. 2020, 11, 3263.)
总结
四川大学余达刚教授课题组首次实现通过可见光参与的光氧化还原催化吲哚与二氧化碳之间的还原去芳构化芳基羧基化方法学。该方法学具有高度的化学选择性,能够避免芳卤的本位交叉偶联反应以及β-H消除副反应的发生;此外,该反应的反应条件温和、官能团兼容性较好、底物范围较广,能够为为常规方法较难合成的吲哚啉-3-羧酸类分子的构建提供了一种行之有效的途径。同时,作者对反应机理进行了深入研究。
参考文献
- [1] a) L. Zhao, Z. Li, L. Chang, J. Xu, H. Yao, X. Wu. Org.Lett. 2012, 14, 2066. DOI: 10.1021/o l300584m. b) X. Li , B. Zhou, R. Yang , F. Yang , R. Liang, R. Liu, Y. Jia. Am. Chem. Soc.2018, 140, 13945. DOI: 10.1021/jacs.8b09186.
- [2] M. J. Krische, Metal Catalyzed Reductive C–C Bond Formation (Springer, 2007). DOI: 10.1007/978-3-540-72879-5.
- [3] D. J. Weix, Acc. Chem. Res. 2015, 48, 1767. DOI:10.1021/acs.accounts.5b00057.
- [4] a) S. Sun, Y. Duan, R. Mega, R. Somerville, R. Martin. Angew. Chem. Int. Ed. 2020, 59, 4370. DOI: 10.1002/anie.201916279. b) X. Zhao, H. Tu , L. Guo, S. Zhu, F. Qing, L. Chu. Commun. 2018, 9, 3488. DOI: 10.1038/s41467-018-05951-6. c) D. Anthony, Q. Lin, J. Baudet, T. Diao. Chem. Int. Ed. 2019, 58, 3198. DOI: 10.1002/anie.201900228.
- [5] K. Wang, Z. Ding , Z. Zhou, W. Kong. J. Am. Chem. Soc. 2018, 140, 12364. DOI: 10.1021/ja cs.8b08190.
- [6] C. Zhuo, C. Zheng, S. You. Acc. Chem. Res.2014, 47, 2558. DOI: 10.1021/ar500167f.
- [7] A. Cerveri, M. Bandini. Chin. J. Chem. 2020, 38, 287. DOI: 10.1002/cjoc.201900446.
- [8] a) R. Liu, Y. Wang, Y. Li, B. Huang, R. Liang, Y. Jia. Angew. Chem. Int. Ed. 2017, 56, 7475. DOI: 10.1002/anie.201703833. b) A. Marchese , F. Lind, Á. Mahon , H. Yoon, M. Lautens. Chem. Int. Ed. 2019, 58, 5095. DOI: 10.1002/anie.201900659.
- [9] X. Qin, M. Lee, J. Zhou. Angew. Chem. Int. Ed. 2017, 56, 12723. DOI:10.1002/anie.201707134.
- [10] a) T. Fujihara, K. Nogi, T. Xu, J. Terao, Y. Tsuji, J. Am. Chem. Soc. 2012, 134, 9106. DOI: 10.1021/ja303514b. b) Q. Meng., S. Wang., B. König. Chem. Int. Ed. 2017, 56, 13426. DOI: 10.1002/anie.201706724. c) Y. Higuchi, T. Mita, Y. Sato. Lett. 2017, 19, 2710. DOI: 10.1021/acs.orglett.7b01055.
- [11] a) V. Yatham, Y. Shen , R. Martin. Angew.Chem. Int. Ed. 2017, 56, 10915. DOI:10.1002/anie.201706263. b) Ye, M. Miao, H. Huang, S. Yan , Z. Yin, W. Zhou, D. Yu. Angew. Chem. Int. Ed. 2017, 56, 15416. DOI:10.1002/anie.201707862. c) Hou, A. Ee, H. Cao, H. Ong, J. Xu, J. Wu. Angew. Chem. Int. Ed. 2018, 57, 17220. DOI:10.1002/anie.201811266 d) Fu, Z. Bo, J. Ye, T. Ju, H. Huang, L. Liao., D. Yu. Nat. Commun. 2019, 10, 3592. DOI:10.1038/s41467-019-11528-8.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!











No comments yet.