
作者:石油醚
导读:
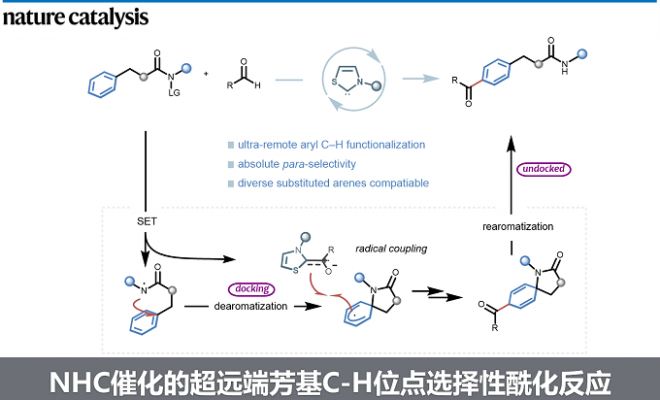
近日,成都大学李俊龙课题组在Nature Catalysis上发表了题为“Remote site-selective arene C–H functionalization enabled by N-heterocyclic carbene organocatalysis”的研究论文。本研究利用氮杂环卡宾有机小分子催化,开发了一种“芳环的超远端”位点选择性的酰基化策略,实现了超远端芳基C(sp2)-H键的位点选择性活化。
“Remote site-selective arene C–H functionalization enabled by N-heterocyclic carbene organocatalysis.
Qing-Zhu Li, Wen-Lin Zou, Zhao-Yuan Yu, Xin-Xin Kou, Yan-Qing Liu, Xiang Zhang, Yu He & Jun-Long Li*
Nat. Catal. 2024. Doi: 10.1038/s41929-024-01194-5”
正文:
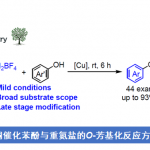
高选择性的远端惰性C-H键官能化一直是有机合成领域的重要挑战之一。除了远端C-H金属化[1-3]、过渡金属催化的σ键活化[4-5]和卡宾或氮烯插入[6]外,自由基介导的转化因其反应条件温和且环保,在远端功能化方面赢得了越来越多的关注。其中,利用自由基介导的分子内氢原子转移(HAT)化学可以实现远端C-H活化,然而这一策略通常局限于距离反应中心不超过7个化学键的C(Sp3)-H键(Fig. 1a-b)。对于远距离的芳基C(sp2)-H键来说,其具有较高的键能、空间位置不利等多种挑战,因而无法通过传统的自由基化学对其进行选择性活化。这里,成都大学的李俊龙课题组利用NHC自由基催化完成了一系列远端芳基C(sp2)–H键的活化,实现了一种全新的“芳环超远端的位点选择性酰基化”反应,为超远端芳基C(sp2)–H键的官能化提供了一个新颖的、绿色的、高效的合成工具(Fig. 1d)。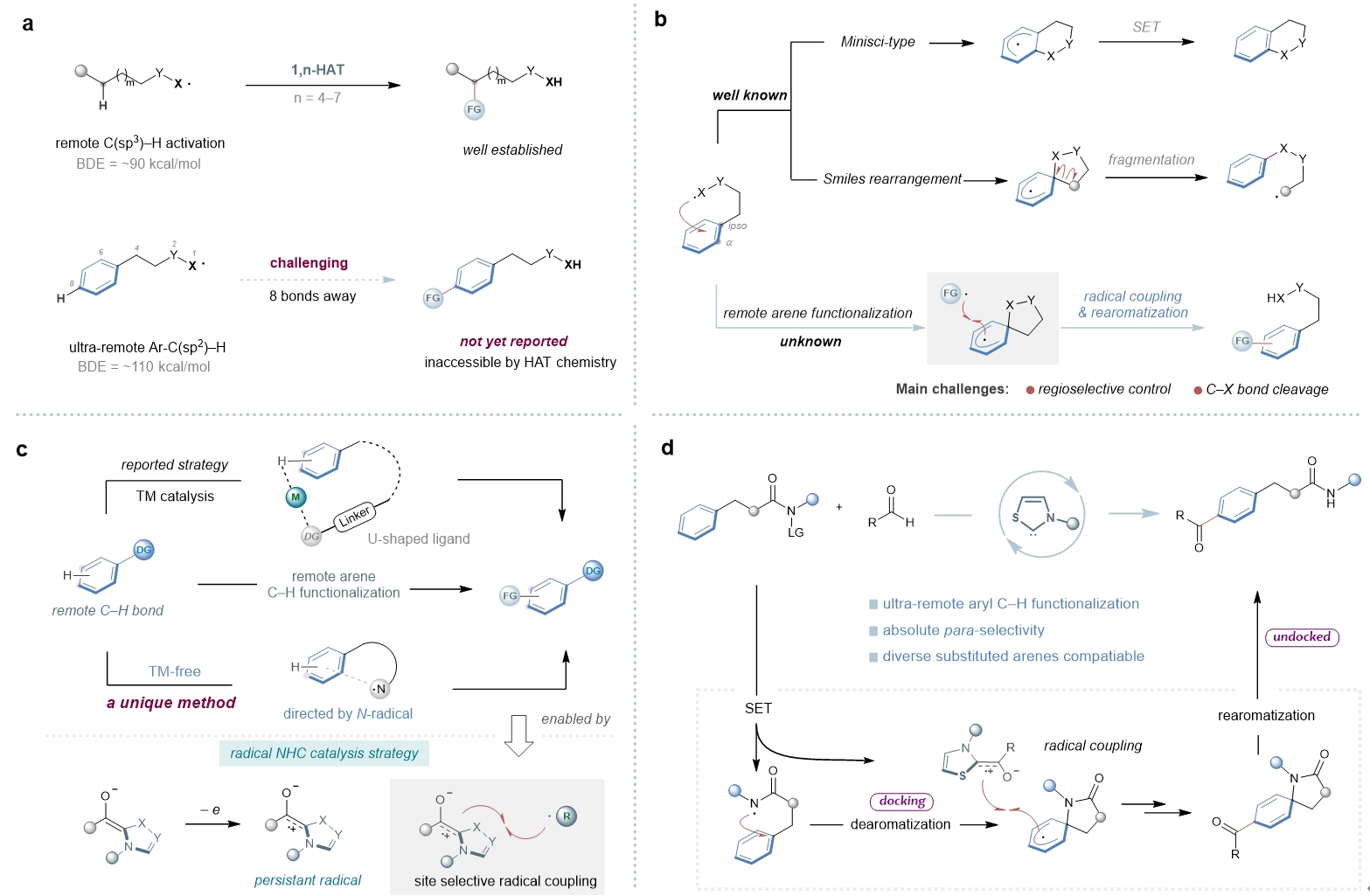
Fig. 1. Background and discovery of N-radical-directed remote arene C–H functionalization. a, Radical-mediated remote C–H functionalization. b, Diverse pathways driven by intramolecular radical addition to arenes. c, Approaches to site selective functionalization of remote arene C–H bonds. d, This work: N-radical-directed para-selective acylation of ultraremote arene C–H bonds. FG, functional group; DG, directing group; TM, transition metal.
作者首先以苯丙酰胺1a和苯丙醛2a为模型底物,通过大量的条件筛选表明:采用氮杂环卡宾N1为催化剂,K3PO4作碱,在甲苯中60℃反应效果最佳,能以96%的产率得到远端酰基化的产物3a(Fig. 2a)。反应条件的敏感性评估结果表明:该反应对各种外部条件都比较耐受,但对氧气较为敏感,因而需要在惰性气体中进行反应(Fig. 2b)。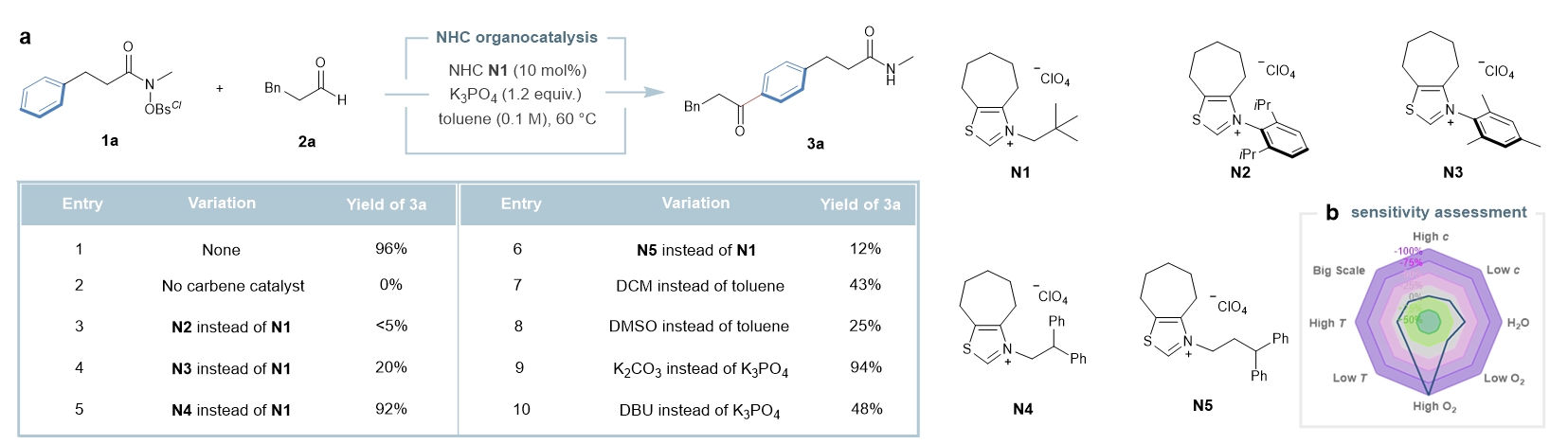 2. Reaction condition assessment. a, Optimal reaction conditions: amide 1a (0.10 mmol), aldehyde 2a (0.25 mmol), NHC (10 mol%) and base (0.12 mmol) in 1 ml of solvent at 60 °C for 12 h; isolated yield. The results of various screening conditions are presented. b, Sensitivity assessment. The robustness and reproducibility of this catalytic method was evaluated by a sensitivity assessment for the reaction conditions.
2. Reaction condition assessment. a, Optimal reaction conditions: amide 1a (0.10 mmol), aldehyde 2a (0.25 mmol), NHC (10 mol%) and base (0.12 mmol) in 1 ml of solvent at 60 °C for 12 h; isolated yield. The results of various screening conditions are presented. b, Sensitivity assessment. The robustness and reproducibility of this catalytic method was evaluated by a sensitivity assessment for the reaction conditions.
令人欣慰的是,该反应策略具有广泛的底物普适性。就醛类底物而言(Fig. 3),不同电性基团取代的苯丙醛、直链脂肪醛、支链烷基醛、含有烯烃官能团或杂原子的烷基醛、环状烷基醛以及乙醛酸乙酯都能顺利进行反应;在对位、间位或邻位上具有吸电子或给电子取代基的苯甲醛、双取代芳醛、稠环芳醛和各种杂芳香醛也都与该催化体系兼容。
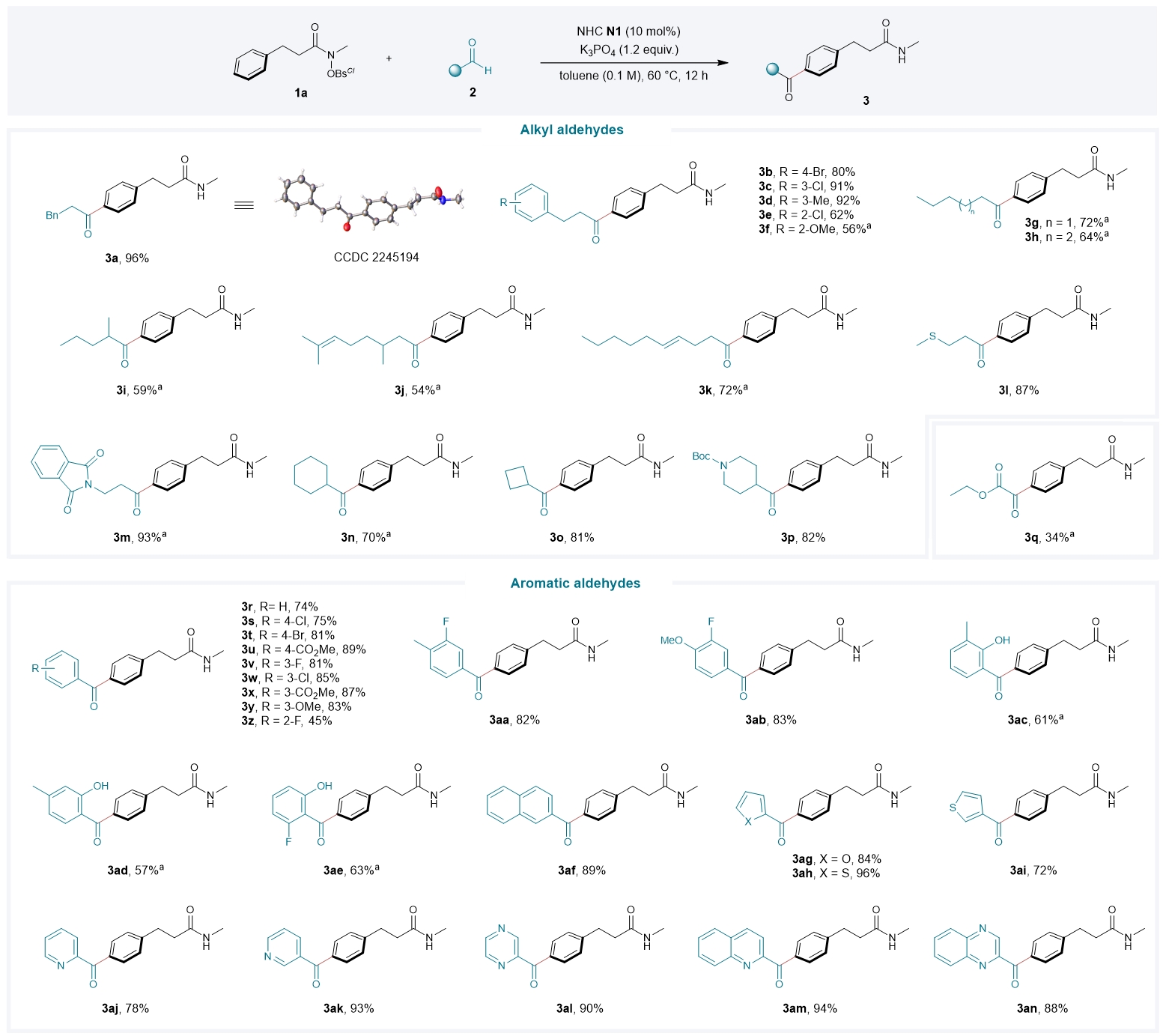
3. Aldehyde scope of the organocatalytic remote acylation. Diverse alkyl and aryl aldehydes were well tolerated in the organocatalytic acylation. Reactions were carried out with amide 1a (0.10 mmol), aldehyde 2 (0.25 mmol), NHC N1 (10 mol%) and K3PO4 (0.12 mmol) in 1 ml of toluene at 60 °C for 12 h; isolated yield; no regioisomer was detected in all cases. a DMSO (0.1 M) was used as solvent, and phenylpropionic acid (0.06 mmol) was used as an additive.
多种类型的酰胺类化合物都适用于该催化反应(Fig. 4)。烷基酰胺底物无论芳环上有不同电性取代基、强定向基、强拉电子基或多个取代基,还是具有稠芳环、支链或增大N-自由基中心的空间位阻对催化效率均没有太大的影响;不同类型的氨基甲酸酯类底物也可以顺利进行反应;含有N-3,3-二苯丙基的氨基甲酸酯底物只得到了苄醇芳基对位酰化的产物,进一步证实了该反应的高位点选择性。此外,该反应还能实现联苯酰胺芳烃底物的位点选择性的远端碳氢官能化。
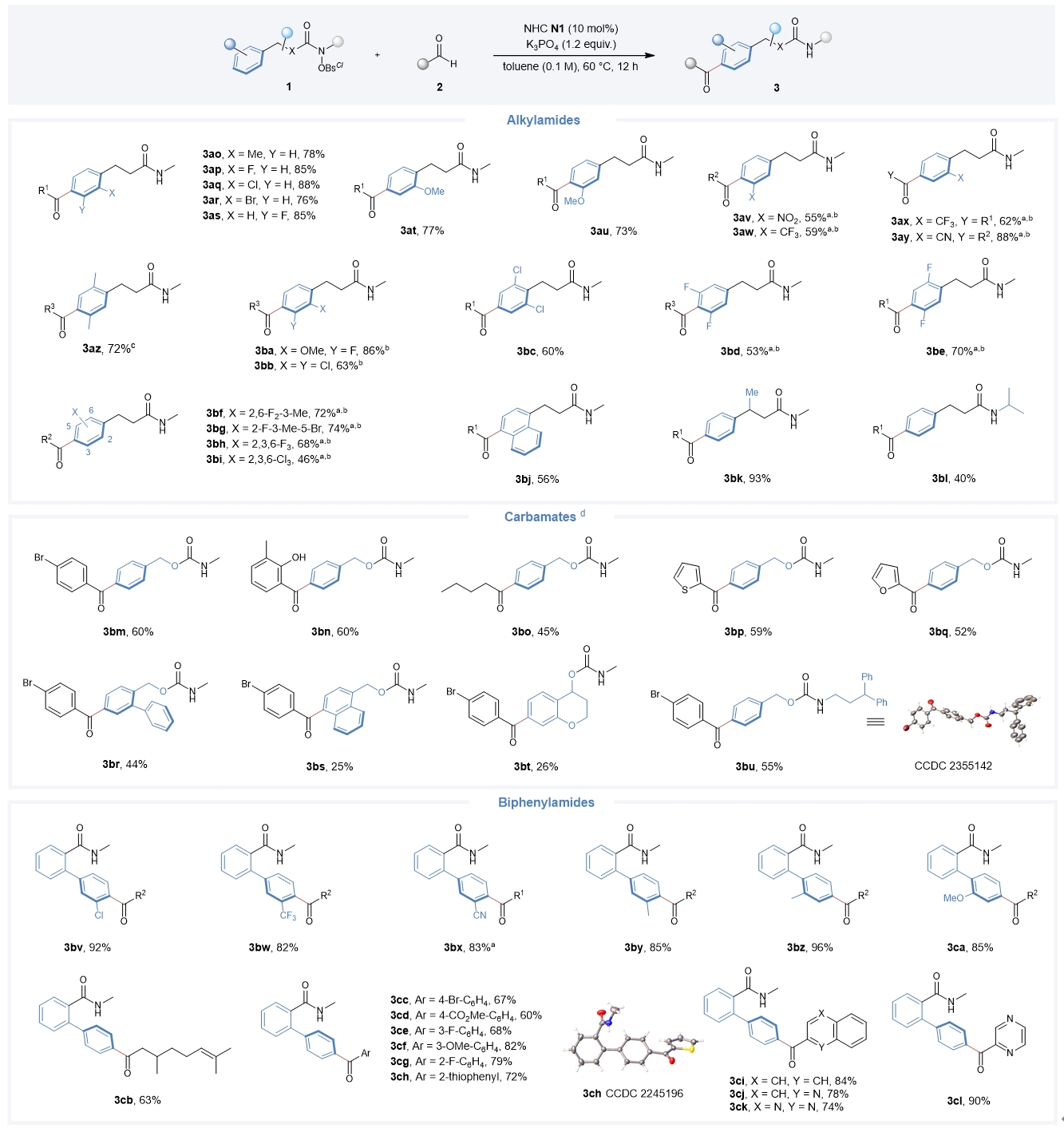 4. Amide scope of the organocatalytic remote acylation. Broad substrate scope of amides was observed in the organocatalytic acylation. See Fig. 2 for reaction conditions; isolated yield; no regioisomer was detected in all cases. a KHCO3 was used as the base. b DCM was used as the solvent. c DMSO (0.1 M) was used, and phenylpropionic acid (0.06 mmol) was used as an additive. d For the carbamate substrates, N2 (10 mol%) was used as the catalyst and DCM (0.1 M) was used as the solvent. R1 = phenylethyl; R2 = pyrazinyl; R3 = 4-Br–C6H4.
4. Amide scope of the organocatalytic remote acylation. Broad substrate scope of amides was observed in the organocatalytic acylation. See Fig. 2 for reaction conditions; isolated yield; no regioisomer was detected in all cases. a KHCO3 was used as the base. b DCM was used as the solvent. c DMSO (0.1 M) was used, and phenylpropionic acid (0.06 mmol) was used as an additive. d For the carbamate substrates, N2 (10 mol%) was used as the catalyst and DCM (0.1 M) was used as the solvent. R1 = phenylethyl; R2 = pyrazinyl; R3 = 4-Br–C6H4.
本文建立的NHC有机催化方案可进一步应用于多种药物骨架、生物活性分子和糖类化合物的官能团化修饰(Fig. 5),如头孢类抗菌药物(cefoxitin)、非甾体类抗炎药芬布芬(fenbufen)和吲哚美辛(indometacin)、治疗痛风药物非布索坦(febuxostat)和丙磺舒(probenecid)、血脂调节药吉非罗齐(gemfibrozil)、治疗痤疮类药物阿达帕林(adapalene)、薄荷醇(L-menthol)、diacetone-D-galactose等。
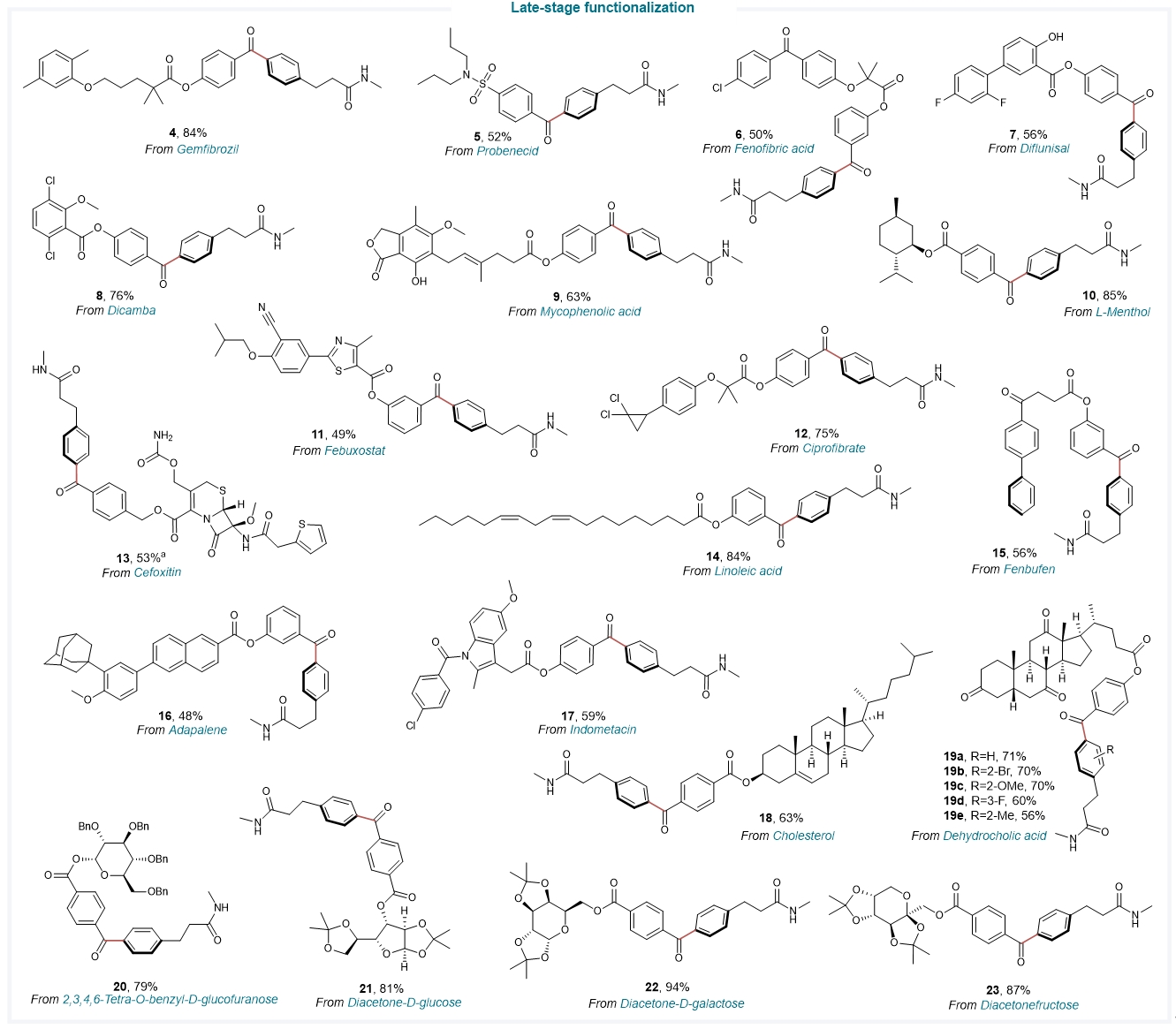
Fig. 5. Late-stage functionalization of drugs and biologically active molecules. The NHC organocatalytic arene C–H acylation protocol was successfully applied to the late-stage functionalization of various pharmaceutical skeletons. See Fig. 2 for reaction conditions. a For compound 11, DCM (0.1 M) was used as the solvent.
随后,该小组通过对苯丙氨酸衍生物的功能化,进一步证明了该催化体系的通用性(Fig. 6)。此外,该反应对苯丙氨酸衍生的多种二肽、三肽、四肽和五肽衍生物也具有良好的兼容性,通过该反应,还能实现糖和多肽骨架的快速连接。
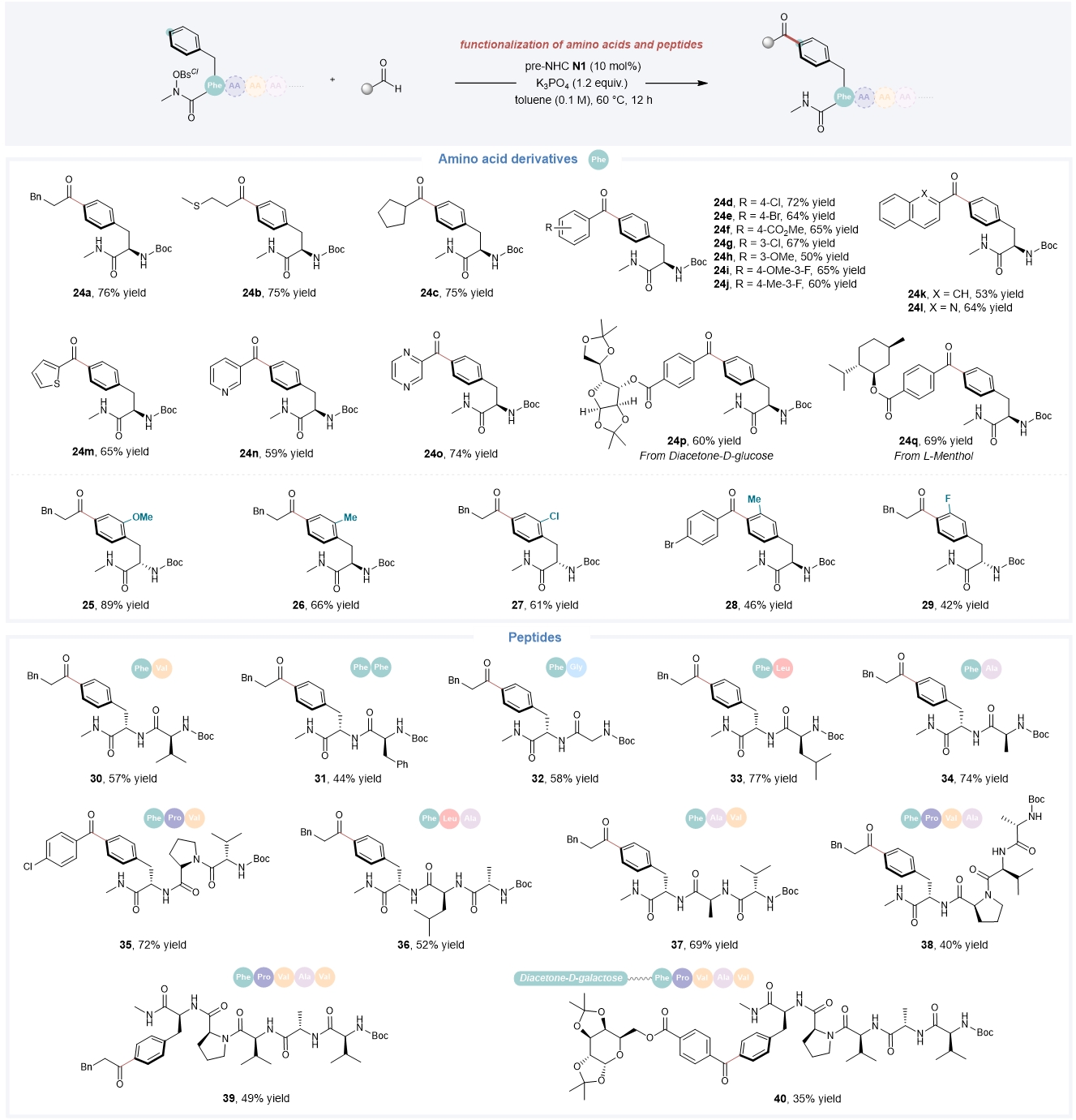
Fig. 6. Site-selective functionalization of amino acid derivatives and peptides. This organocatalytic approach is suitable for the functionalization of amino acids and peptides. See Fig. 2 for reaction conditions.
该NHC催化自由基远端碳氢键的官能化能够实现克级规模放大,得到的酰化产物还可以进行多样化的化学转化(Fig. 7a),进一步彰显了本方法的实用性。为了阐明该远程酰化的反应机理,作者进行了详细的机理实验研究(Fig. 7b)。首先,自由基抑制实验和自由基钟实验表明该催化反应涉及环己二烯自由基中间体;当9-蒽底物参与反应时,作者可以得到螺环酮中间产物,进一步加入强碱处理便可以顺利得到对应的酰化产物,表明该反应确实存在去芳构化的螺环酮中间体。此外,芳烃对位封堵或不饱和酰胺底物为反式烯烃时,反应都不能发生。氘代实验结果表明去质子化过程不是该反应的决速步骤。
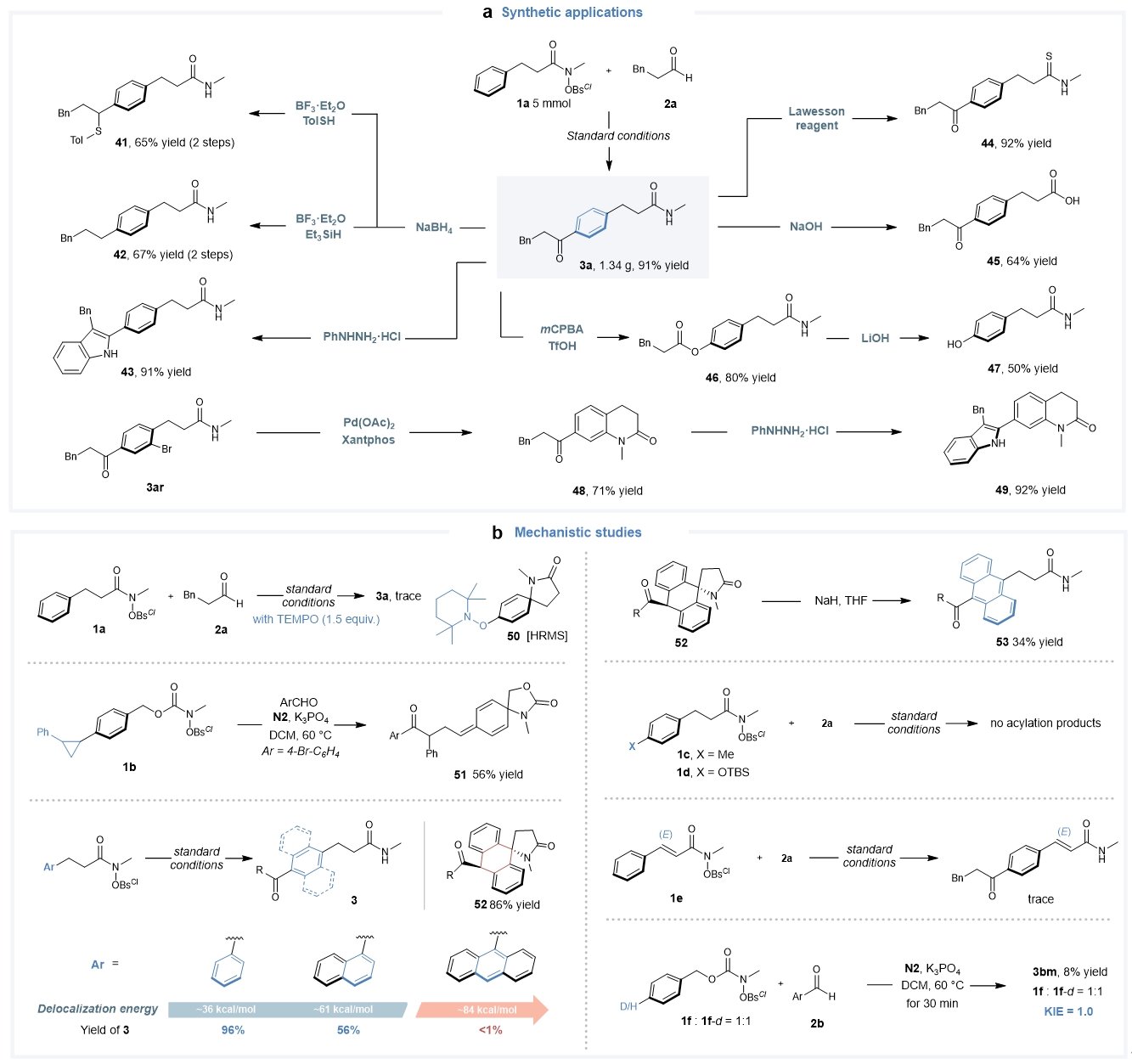 Fig. 7. Further experimental studies. a, Synthetic applications. b, Experimental investigations on reaction mechanism.
Fig. 7. Further experimental studies. a, Synthetic applications. b, Experimental investigations on reaction mechanism.
在以上实验结果的基础上,作者进一步通过密度泛函理论(DFT)计算,研究了各反应路径的能量,通过对自旋密度分布、扭曲和相互作用等分析,为NHC自由基催化远端碳氢键活化的位点选择性提供了合理的解释(Fig. 8)。在此基础上,作者提出了合理的反应机理。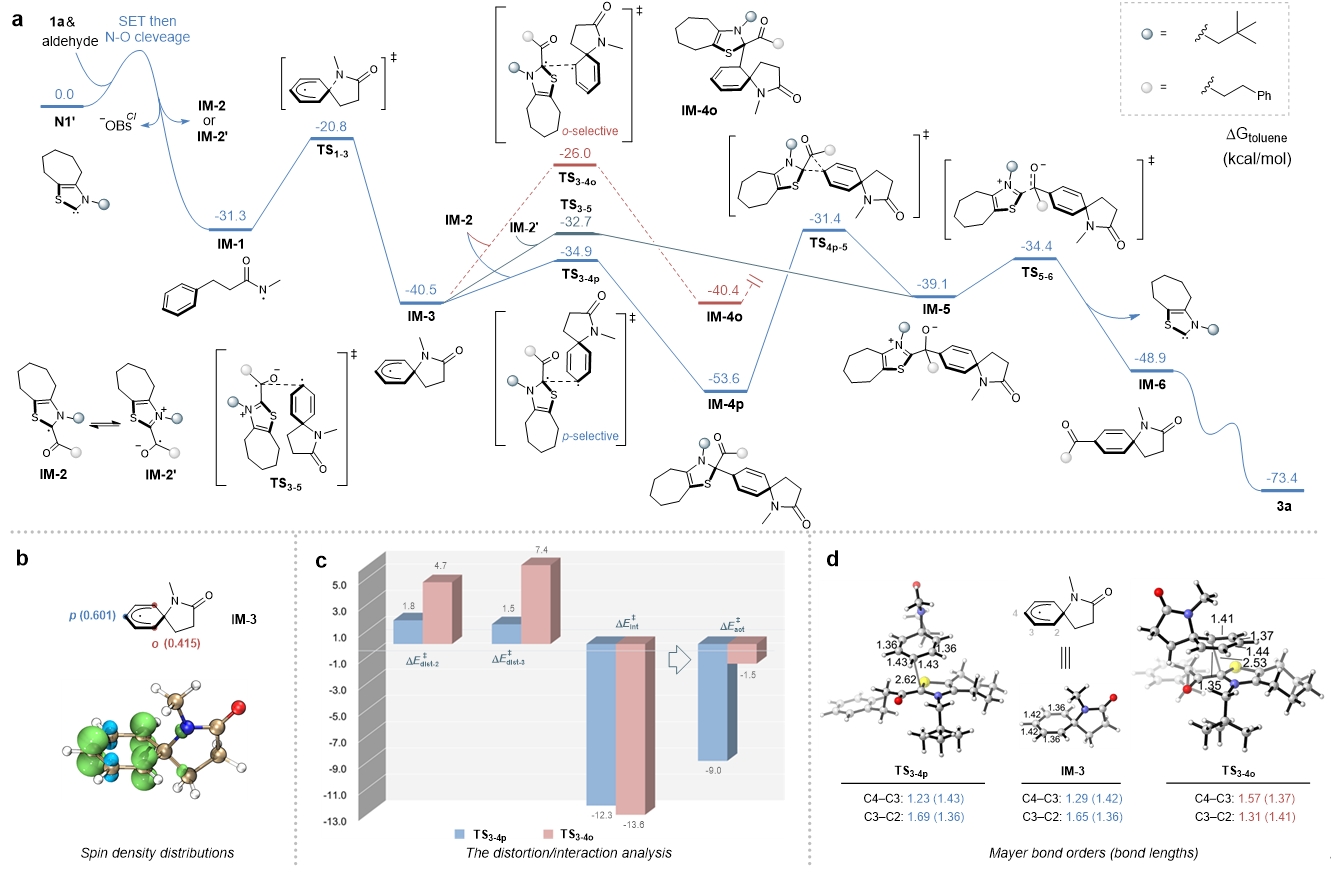
Fig. 8. DFT calculations. The energy profiles of proposed reaction pathways were investigated by DFT calculations. The regioselectivity was further rationalized by analysis of spin density distribution, distortion and interaction, as well as the Mayer bond orders. a, Energy profiles for the reaction pathways. b, Spin density distribution. c, Energies (kcal mol−1) of distortion and interaction. d, Mayer bond orders and C–C bond lengths (Å).
总结:
成都大学李俊龙课题组开发了一种“芳环超远端的位点选择性酰基化”反应,实现了距离活泼位点8根化学键的远程C–H键的高选择性活化。同时,该催化体系可以克服芳烃固有的电性效应和位阻效应,完成了一系列传统方法难以实现的酰基化反应,拓展了NHC自由基催化的应用范围。该催化体系为实现超远端芳基碳氢键的官能化提供了一个绿色简单的方法。
(李俊龙团队供稿)
参考文献:
- [1] Z. Fan, X. Chen, K. Tanaka, H. S. Park, N. Y. S. Lam, J. J. Wong, K. N. Houk, J.-Q. Yu. Nature 2022, 610, 87. doi: 10.1038/s41586-022-05175-1.
- [2] N. Y. S. Lam, Z. Fan, K. Wu, H. S. Park, S. Y. Shim, D. A. Strassfeld, J.-Q. Yu, J. Am. Chem. Soc. 2022, 144, 2793. doi: 10.1021/jacs.1c12654.
- [3] J.-J. Li, J.-H. Zhao, H.-C. Shen, K. Wu, X. Kuang, P. Wang, J.-Q. Yu. Chem 2023, 9, 1452. doi: 10.1016/j.chempr.2023.04.003.
- [4] J. A. Leitch, C. G. Frost, Chem. Soc. Rev. 2017, 46, 7145. doi: 10.1039/c7cs00496f.
- [5] N. Hofmann, L. Ackermann, J. Am. Chem. Soc. 2013, 135, 5877. doi: 10.1021/ja401466y.
- [6] H. M. L. Davies, J. R. Manning, Nature 2008, 451, 417–424. doi: 10.1038/nature06485.

















No comments yet.