
本文作者:杉杉
导读
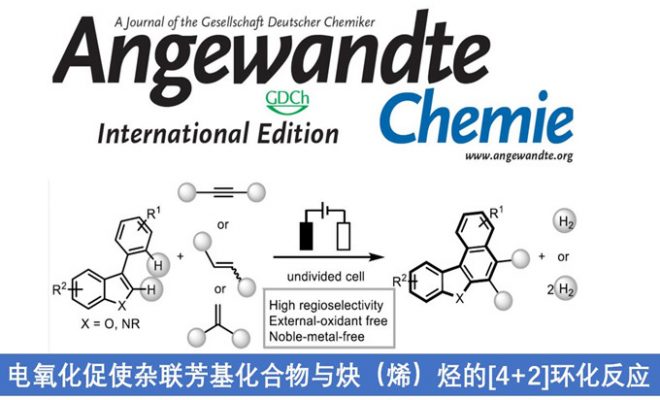
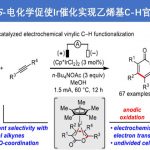
稠合芳基化合物由于具有独特的电子特性以及生物活性的多样性,从而在材料和药物领域被广泛的研究。近日,武汉大学雷爱文教授课题组在德国应化杂志(Angewandte Chemie-International Edition)发表论文,在电化学氧化的条件下,实现杂联芳基化合物与炔烃或烯烃的[4+2]环化反应,获得高收率和优异区域选择性稠合杂芳基化合物。该方法避免了外部氧化剂的使用以及原料预官能化的要求。此外,原位产生杂二芳基自由基阳离子作为关键的中间体。
Electrochemical Oxidative [4+2] Annulation for the π-Extension of Unfunctionalized Hetero-biaryl Compounds
Xia Hu, Lei Nie, Guoting Zhang and Aiwen Lei *
Angew. Chem. Int. Ed. ASAP DOI: 10.1002/anie.202003656
正文
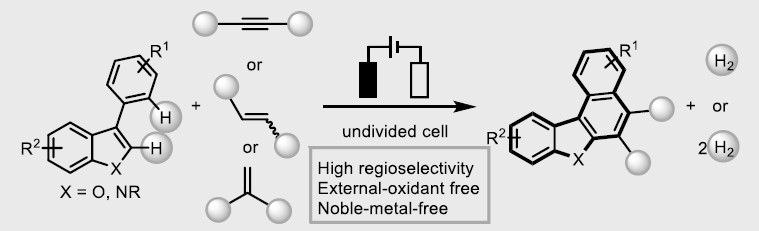
稠合多环杂芳基化合物由于具有独特电子特性和生物活性,广泛存在于药物、生物活性分子、光电材料等中。因此,对于构建多环杂芳基化合物的有效方法,已在有机化学和材料科学中引起了极大的关注。经典的反应常常涉及卤化、交叉偶联、环化和氧化等多步过程。相反的,过渡金属催化π-共轭试剂与(杂)芳烃的环化反应(APEX),作为快速构建稠合多环杂芳基化合物高效方法。通常,杂芳烃的双C-H芳基化需要对联芳基化合物预氧化,如二芳基碘鎓盐、二碘二联芳基(Scheme 1A)。此外,涉及炔烃或烯烃作为π-共轭体系的 [2+2+2]环加成反应,通常也受底物范围的限制,特别对于内部炔烃或电子不足的烯烃(Scheme 1A)。在此,武汉大学雷爱文教授课题组实现了杂二联芳基化合物与炔烃或烯烃的[4+2]环化反应,涉及自由基阳离子途径。此外,通过恒定电流电解过程,无需化学氧化剂和杂联芳基或π-共轭试剂的预功能化处理(Scheme 1B)。
不饱和化合物通过单电子氧化形成自由基阳离子中间体(亲电子中间体),可与亲核试剂反应或进行周环反应。此外,氧化偶联反应涉及烯烃或芳烃自由基阳离子,因此,作者设想是否可以使用杂二芳基自由基阳离子中间体,实现杂二芳基化合物和炔烃或烯烃的[4+2]环化反应。作者假设,由(杂)芳烃的单电子氧化产生的联芳基自由基阳离子I,经炔烃或烯烃的亲和进攻击形成二硅基自由基阳离子II。更重要的是,由于杂原子的极化作用,可很好地控制这种亲核进攻的区域选择性。随后,经去质子化和环化产生中间体III,再经进一步氧化芳构化从而获得多环杂芳族化合物。同时,阴极还原产生H2(Scheme 1B)。

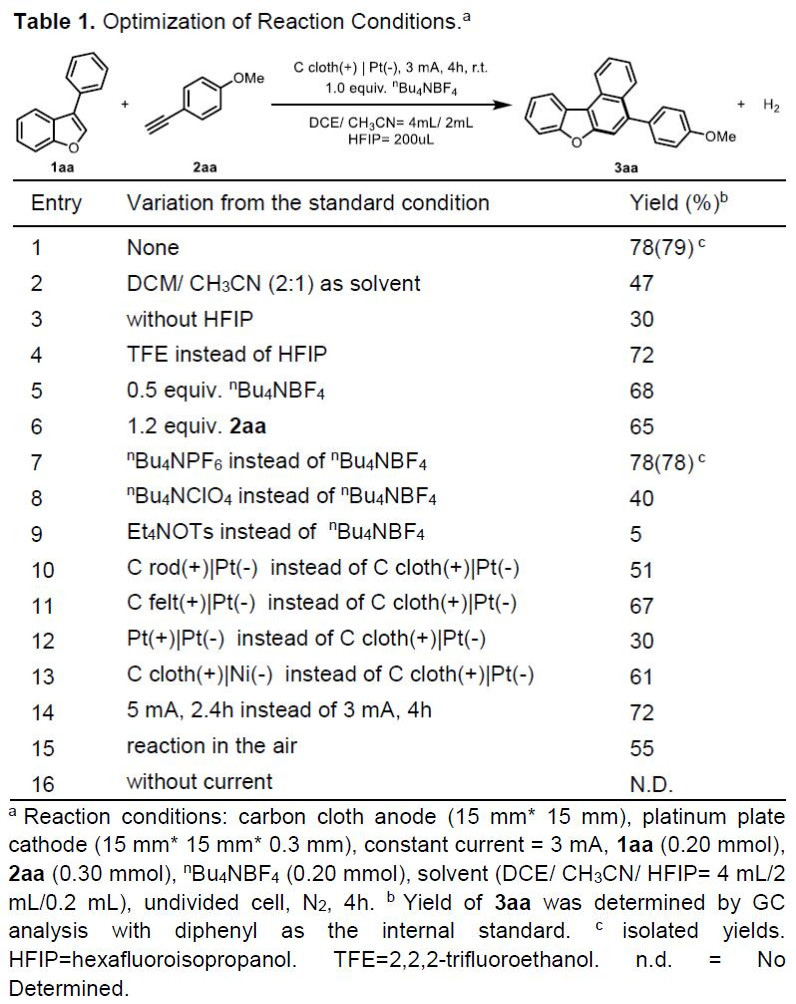
首先,作者以3-苯基苯并呋喃1aa和4-乙炔基苯甲醚2aa作为模型底物,进行了相关[4+2]环化反应条件的筛选(Table 1)。经过大量的筛选之后,作者发现,当在碳阳极和铂阴极的无隔膜电解槽中,在惰性气氛保护下,于DCE/MeCN/HFIP的混合溶剂以3 mA室温反应4h,即可获得79%收率的产物3aa(entry 1)。而使用DCM/MeCN=2:1混合溶剂,收率明显下降(entry 2)。同时,含氟的助溶剂对于转化至关重要,它可以稳定自由基阳离子。如在没有HFIP时,3aa的收率降至30%(entry 3)。而与六氟异丙醇相比,三氟乙醇具有相似的反应活性(entry 4)。此外,电解质或炔烃2aa的减少,收率会有所降低(entries 5-6)。若将电解质更改为nBu4NPF6也具有良好的反应性,但使用nBu4NClO4和Et4NOT时,反应效率明显下降(entries 7-9)。而对电极材料的筛选中,如碳棒阳极、碳毡阳极、铂板阳极或镍板阴极,均没有提高收率(entries 10-13)。而将电流增加到5 mA时,产率较低(entry 14)。此外,在无氮气保护时,环化反应仍能顺利进行,但收率降低(entry 15),若无电流时则不发生反应(entry 16)。
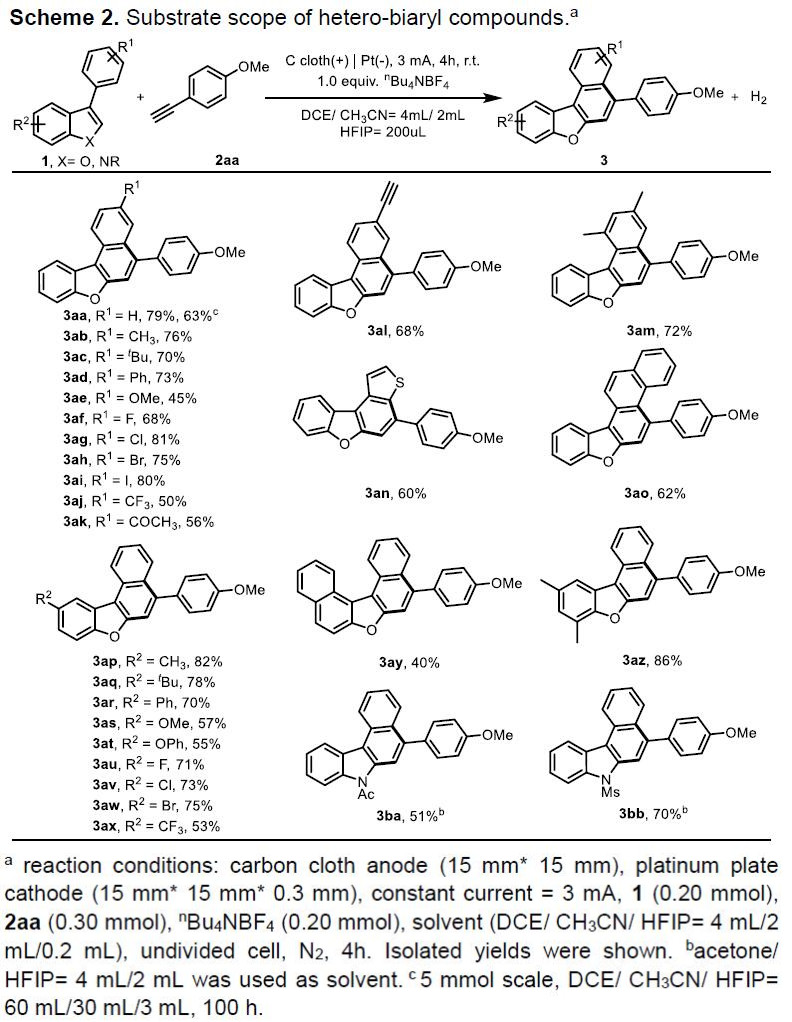
在获得上述最佳反应条件后,作者固定4-乙炔基苯甲醚2aa,对杂联芳基化合物底物1进行了扩展(Scheme 2)。反应结果表明,在3-芳基苯并呋喃的R1取代含有给电子基团(-Me,-tBu,-Ph,-OMe)、卤素(-F,-Cl,-Br,-I)和吸电子(-CF3,-Ac)基团时,均可与2aa反应,生成相应产物3aa–3ak。当以3-(4-乙炔基苯基)苯并呋喃作为底物时,也可获得具有高化学和区域选择性的环化产物3a1。当苯基的邻、对位被甲基取代时,可获得相应的产物3am。同样,杂环取代的底物(如萘基、噻吩基),也与体系兼容,从而获得具有区域选择性的环状产物3an和3ao。随后,在对3-芳基苯并呋喃的R2取代研究时发现,反应不受电子效应影响,均可获得相应的环化产物3ap–3ax。当使用1-苯基萘[2,1-b]呋喃作为底物时,可获得多环芳烃产物3ay。5,7-二甲基-3-苯基苯并呋喃同样与体系兼容,获得产物3az。此外,由于具有给电子基团产物(3ae,3as,3at,3ay)具有相对较低氧化电位,易发生过度氧化,从而导致收率偏低。同时,由于具有缺电子取代的基底物不完全转化,从而导致产物(3aj,3ak,3ax)的收率也偏低。此外,吲哚衍生物也适用于该催化体系,乙酰基或甲基磺酰基保护的3-苯基吲哚,可在丙酮/HFIP混合溶剂中,获得相应的含氮多环杂芳基化合物3ba和3bb。
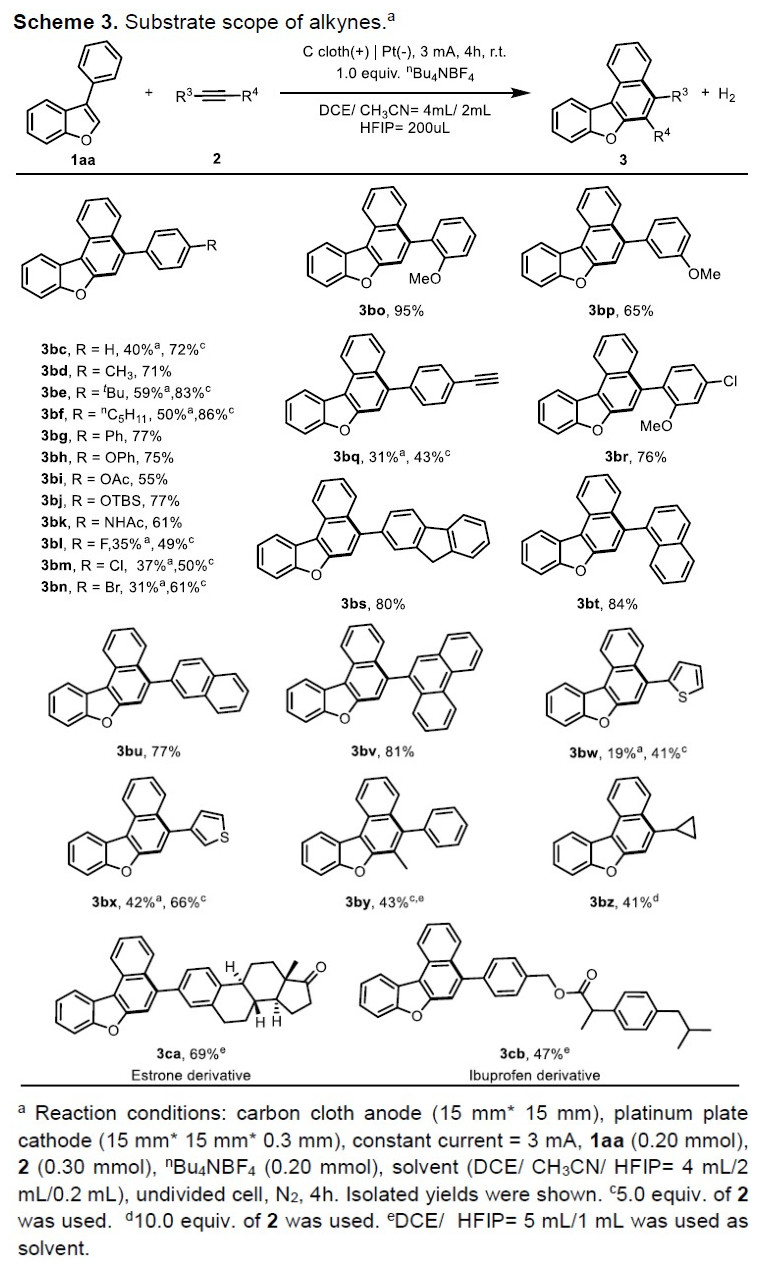
随后,作者固定3-芳基苯并呋喃底物1aa,对炔烃2的反应范围进行了研究(Scheme 3)。反应结果表明,在标准反应条件下,使用无取代苯基乙炔底物时,可获得40%收率的产物3bc,若将苯乙炔增加至5当量时,收率可提高至72%。带有给电子基团(-Me,-tBu,-nC5H11,-Ph,-OPh,-OAc,-OTBS,-NHAc)的苯乙炔衍生物均可与1aa反应,获得相应的产物3bd–3bk。同样,含有卤素官能团的炔烃底物也与体系兼容,从而获得产物3bl–3bn,为后期修饰提供了多种可能。具有邻、间位含有甲氧基取代的苯基乙炔底物,也与体系兼容,获得相应的产物3bo和3bp。而使用1,4-二乙炔基苯时,获得单环化产物3bq。当将4-氯-1-乙炔基-2-甲氧基苯用作亲二烯体时,可获得76%收率的产物3br。此外,一些其它的芳基、杂芳基炔烃(如萘、芴、基菲、噻吩等),均可获得相应的产物3bs–3bx。同时,内部炔烃也可获得具有较高区域选择性环合产物3by。通过反应条件的优化,脂族炔烃(乙炔基环丙烷)也成功地以41%收率获得所需的产物3bz。值得注意的是,对雌酮(3ca)和布洛芬衍生的炔烃(3cb)的后期修饰,进一步证明了反应的实用性。
紧接着,作者将底物范围扩展到芳基烯烃(Scheme 4)。反应结果表明,烯烃具有更高的反应性。当苯乙烯对位上不受电子效应和定位效应的影响,均可获得在相应的产物5aa–5ak。而与2-乙炔基萘(5a1)相比,2-乙烯基萘的反应性稍差。同时,内部烯烃(如β-甲基苯乙烯)也可生成相应的产物5am。此外,3-苯基苯并呋喃与α-甲基苯乙烯的[4+2]环化后获得产物5an,这是由于重排-氧化芳构化过程所致。同时,通过产物3aa或5aa的克级实验,进一步证明了反应的实用性。

此外,作者发现,苯并呋喃衍生物不仅可以充当4π合成子,同时也可作为亲二烯体。如苯并呋喃可以通过环化和氧化芳构化过程(Scheme 5a)与1,1-二苯乙烯进行偶联获得唯一产物8aa,该芳构化产物不能通过光化学氧化获得。而将3-甲基苯并呋喃用作亲二烯体时,获得了具有高区域选择性的二氢萘并[2,1-b]苯并呋喃得单一非对映异构体产物8ab(Scheme 5b)。同时,产物可通过氧化偶联进一步扩展。如由3ao或3cc经DDQ氧化环化后,即可获得高收率的多环稠合化合物9ao或9cc(Scheme 5c)。

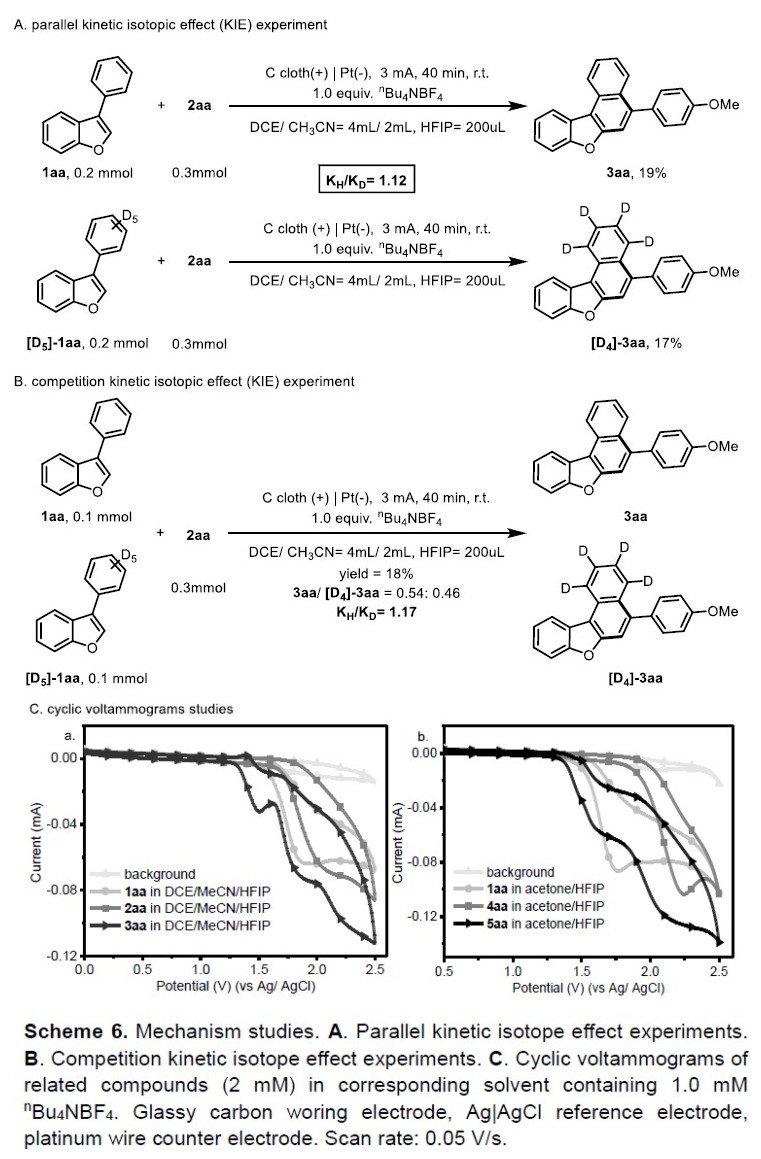
为了进一步了解反应的机理,作者进行了相关的对照实验(Scheme 6)。当使用底物1aa和[D5]-1aa作为二烯时,进行了分子间的动力学同位素效应(KIE)实验,KIE值分别为1.12和1.17 (Scheme 6a-6b)。这些结果表明,第二个芳基碳氢键的断裂可能不是决定步骤。 此外,作者还进行了循环伏安法(CV)实验(Scheme 6c)。从图上可知,1aa的氧化电位低于2aa和4aa,因此,在标准条件下1aa比2aa或4aa更容易被氧化。此外,由于产物3aa或5aa比底物更容易被氧化,从而导致产物的收率与底物的转化不一致。
总结
武汉大学雷爱文教授课题组报道了在电化学氧化的条件下,实现杂联芳基化合物与炔烃或烯烃的[4+2]环化反应,从而获得高收率和优异的区域选择性的稠合杂芳基化合物。该反应具有广泛的底物范围、良好的官能团耐受性、温和的反应条件等优点。同时,杂联芳基化合物的阳极氧化形成杂芳基自由基阳离子,作为实现区域选择性的关键。同时,克级实验以及产物的后期修饰,进一步证明了反应的实用性。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!












No comments yet.