
本文作者:alberto-caeiro
导读
Jacobsen课题组报道了手性脲催化剂催化neryl chloride衍生物的手性尾-头环化反应。实验表明远端的烯烃在氯离子离去中起到了作用,并抑制了消除反应,并对反应速率和对映选择性有贡献。动力学实验和DFT计算都表明该反应中有两分子的手性脲催化剂,并通过过渡态的选择性稳定作用实现了反应的对映选择性。
“Enantioselective Tail-to-Head Cyclizations Catalyzed by Dual-Hydrogen-Bond Donors”
Dennis A. Kutateladze, Daniel A. Strassfeld and Eric N. Jacobsen* J. Am. Chem. Soc. 2020. DOI: 10.1021/jacs.0c02665.
论文介绍
萜类和类萜化合物是一类重要的天然产物,他们在自然界中通常是以异戊二烯为原料,经萜烯环化酶催化合成的。在这些多烯环化中,有两种合成模式(Figure 1A):(1)通过烯烃质子化实现的头-尾环化(Head to tail HT);(2)通过烯丙基焦磷酸酯离去实现的尾-头环化(Tail to head TH)。底物预组织和酶活性位点中的非共价稳定相互作用可用来调节所得碳正离子中间体的反应性,从而实现选择性重排和C-C键生成,最终产生各样的天然产物(Figure 1B)。
Jacobsen教授设想通过使用小分子催化剂模拟酶在反应中对碳正离子中间体反应性的调节实现尾-头环化反应。具有两个氢键的手性脲催化剂已被证实可对通过离去离子的攫取产生的碳正离子有良好的手性控制[1],且具有类似酶的非共价稳定碳正离子的能力。在此,Jacobsen教授实现了手性脲催化neryl chloride衍生物对映选择性尾-头环化反应(Figure 1C)。
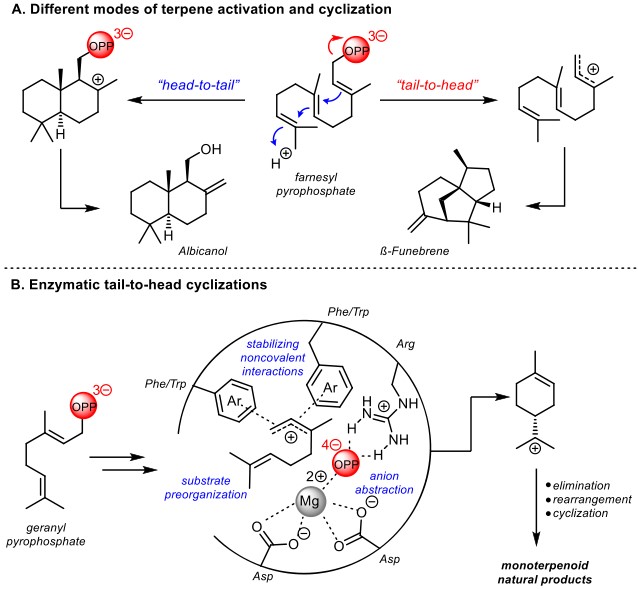
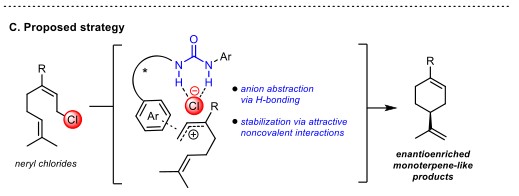
Figure 1. (A) Head-to-tail and tail-to-head cyclization reactions. (B) Schematic illustrating Nature’s strategy for controlled ionization-dependent cyclizations. (C) Proposed strategy for enantioselective tail-to-head cyclizations catalyzed by chiral hydrogen-bond donors.
烯烃的顺反对反应有巨大的影响。反式烯烃geranyl chloride在标准条件下反应很慢,且相当一部分发生消除而非环化产物(Figure 2)。而使用顺式烯烃neryl chloride(1a)时,相同条件下反应有88%的转化,且绝大部分为环化产物。当使用手性脲催化该反应时,在对手性催化剂进行广泛筛选后(see SI for details),最好的结果仍只有34% ee值。
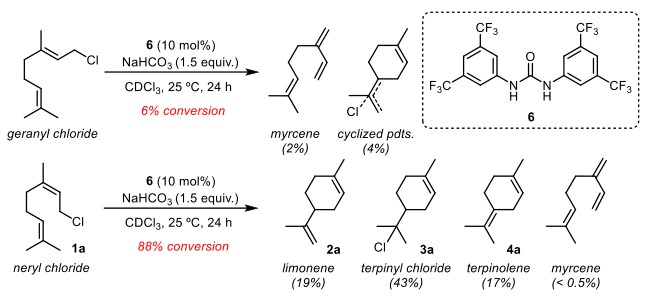
Figure 2. Differing reactivity observed in the urea-catalyzed cyclization of E and Z isomers. Conversions and yields were assessed from crude reaction mixtures using 1H-NMR with mesitylene as an internal quantitative standard.
考虑到1a在不对称反应中是很难的底物,于是他们将C-3位换为苯基进行后续研究。对于底物1b,该反应能有63%的NMR收率和87% ee值,另一手性环化产物3b则有20%收率,86% ee值,类似的ee值与同一反应过渡态有关。非手性环化产物4b和5b的收率分别是12%和5%,其中5b是过渡态经[1,2]-H迁移后消除得到。但使用顺式烯烃2b时,反应速率则变慢(24 h, 7% conv.),且只有50% ee值。
底物范围中(Figure 3),吸电子芳环相较于给电子芳环有更好的反应性和手性控制,邻位有取代基时对反应性和对映选择性也有提高。而使用环己烷作为取代基时,反应性和对映选择性较甲基时有明显的提高。这些说明取代基位阻和电性对反应都有影响。
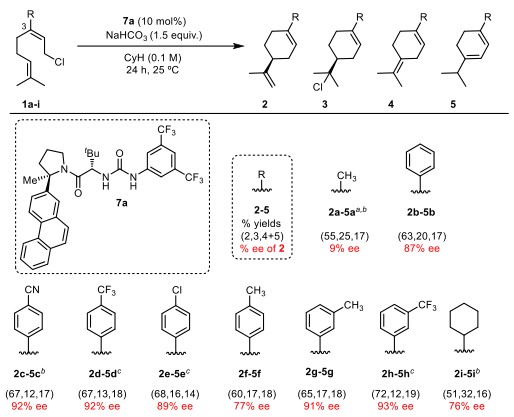
Figure 3. Substrate scope. All reactions were performed on 0.15 mmol scale and proceeded to complete conversion. The ee values are for products 2a-i. Alkyl chlorides 3b, 3h, and 3i were generated in 86% ee, 91% ee, and 70% ee, respectively.
在KIE实验中(Figure 4A),kH/kD = 0.944(3)。此相反的二级同位素效应值表明远端烯烃参与到决速步。0.944的数值可能是由过渡态中烯烃已经部分由sp2杂化至sp3,C-C键也部分形成。阳离子取代基效应的实验中(Figure 4B),ρ=-1.3表明反应经过了碳正离子过渡态。在动力学分析中,反应对底物是一级反应,对碱是零级反应,而对催化剂是1.17,这说明反应过渡态中催化活性物种是手性脲的单体和二聚体的复合物。含有两个氢键的手性脲在液相和固相都可以二聚形式存在,该反应中也是如此(see SI for details)。
根据已有信息,作者推测了如下的反应机理(Figure 4C):氯原子在催化活性物种和远端烯烃的共同作用下离去达到烯烃离子化的反应过渡态,随后烯烃完成进攻得到碳正离子中间体,随后反应得到产物。
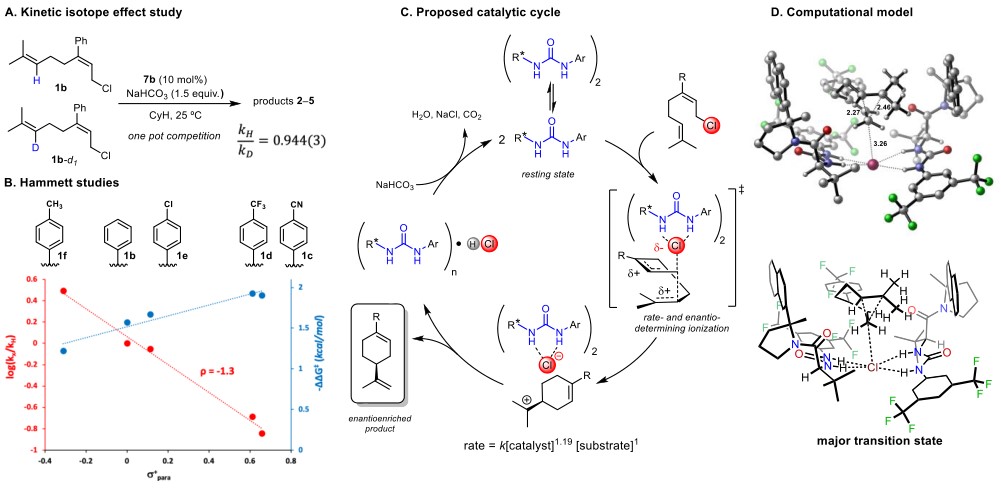
Figure 4. Mechanistic studies. (A) One-pot competition secondary H/D KIE experiment. (B) Hammett studies. In red: Relative rates of reaction of 1b-1f promoted by catalyst 7b. In blue: Enantioselectivities in the formation of 2b-2f promoted by 7a. (C) Proposed catalytic cycle based on the KIE data and the experimentally determined rate law. (D) Transition state model for the pathway leading to the major enantiomeric product in the cyclization of 1d.
后续的DFT计算得到的过固态也表明反应的活性催化物种是手性脲的单体和二聚体的复合物(Figure 4D)。在过渡态中,催化剂上的萘基团与电荷中心靠近以稳定过渡态,从而实现了手性中心的诱导。实验数据也证实了取代基不同对反应选择性有影响(Figure 5)。对于不同的取代基,反应速率和对映选择性有正相关,这说明当催化剂的取代基越能稳定过渡态时,反应速度和对映选择性都会随之提高,这与远端烯烃参与的决速步相符。
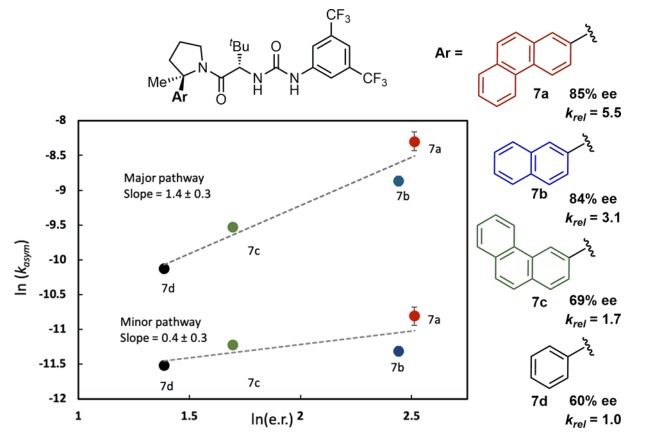
Figure 5. Effect of catalyst aryl substituents on reaction rate and enantioselectivity.
总结
Jacobsen课题组报道了手性脲催化剂催化neryl chloride衍生物的手性尾-头环化反应。远端烯烃协同氯离子离去生成过渡态既是反应决速步也是对映选择性决定步。动力学实验和DFT计算都表明该反应中有两分子的手性脲催化剂,并通过过渡态的选择性稳定作用实现了反应的对映选择性。
参考文献
- [1] a. Jacobsen, E. N. J. Am. Chem. Soc. 2007, 129, 13404. DOI: 10.1021/ja076179w; b. Jacobsen, E. N. ACS Catal. 2016, 6, 4616. DOI: 10.1021/acscatal.6b01384; c. for a review, see: Jacobsen, E. N. Angew. Chem., Int. Ed. 2013, 52, 534. DOI: 10.1002/anie.201205449.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!














No comments yet.