
投稿作者 alberto-caeiro
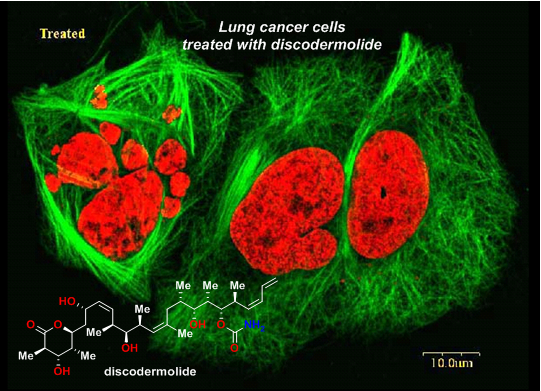
海洋占地球表面的70%左右,其中含有许多有独特生物活性的化学物质。但在许多例子中,海洋天然产物只能分离出很少的量,远远不足以被人工利用。此次的全合成工作就是针对海洋天然产物discodermolide,在Novartis的药学实验室进行的一次百克级的合成工作[1]。
Discodermolide是从大巴哈马群岛(Grand Bahama Island)的一类海绵(discodermia dissoluta)中分离出的聚酮化合物,并于1990年由NMR和X结晶衍射得到其结构为图1所示[2]。虽然人们很早就认识到discodermolide对生长抑制的作用,但其成为明星分子是在人们得知其对双向淋巴细胞反应的抑制作用后(two-way mixed-lymphocyte reaction assay,一类免疫抑制试验),并且其抑制活性在试管试验和活体试验中都得到验证。
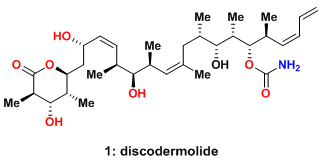
图1:MolecularStructureof Discodermolide
Discodermolide的首次全合成是由Amos B. Smith[3] (University of Pennsylvania)于1995年完成的,其后有另外5个小组(David C. Myles[4] from UCLA; James A. Marshall[5] from University of Virginia; Ian Paterson[6] from Cambridge University; James S. Panek[7] from Boston University; Janick Aedisson[8] from Université de Cergy-Pontoise)完成了全合成工作。但是,S. J. Mickel在Novartis的工作仍然值得在此详细的介绍一番。通过他们的策略,在没有生物发酵的条件下,实现了100g的化学合成,对于这么复杂的天然产物,这无疑是难度巨大,值得被人称赞学习的。遗憾的是,在进行500g级合成时,临床试验发现人们没有预料到的副毒性,合成也随即被叫停。故此次介绍的是其在2004年发表在Organic Process Research & Development上的60g规模的合成工作。
1.逆合成分析
1999年Novartis公司因临床试验需要(100g),开启了化学合成discodermolide的工作。但在开启工作之前,S. J. Mickel领导的小组对已有的合成工作进行研究,希望找到可以大规模合成的有效途径。Discodermolide是一类含有δ内酯环、13个连续手性中心和4个双键的复杂缩酮化合物,其结构相当复杂,大量合成极具难度。从已有报道的全合成路线中我们可以看出,他们都是通过碎片拼接的方式做到最终的合成的,这些碎片的切断位点都在这些双键的周围。(图2)
在Mickel的策略中,分子被解构成9-11三个碎片,他们都含有甲基-羟基-甲基这三个手性中心的碎片。运用这个结构特点,合成家们设计了一个共同的前体,由此制备三个不同的片段。其中有两个很好的前体,一是Weinerb酰胺,二是间位二醇。
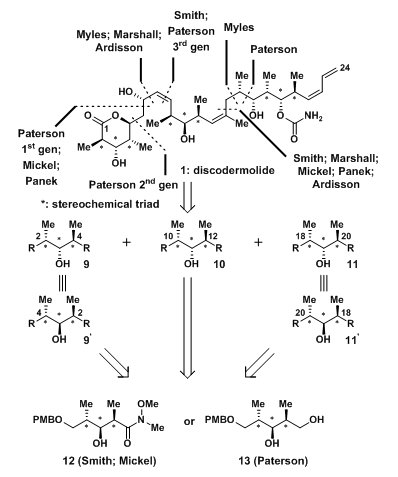
图2:Disconnection Strategies Employed in Total Synthesis of Discodermolide
为了减少反应步数,避免使用特殊的仪器设备(低温、高压等),降低反应时间以及便于分离提纯,Mickel设计了如下的合成路线,图三。
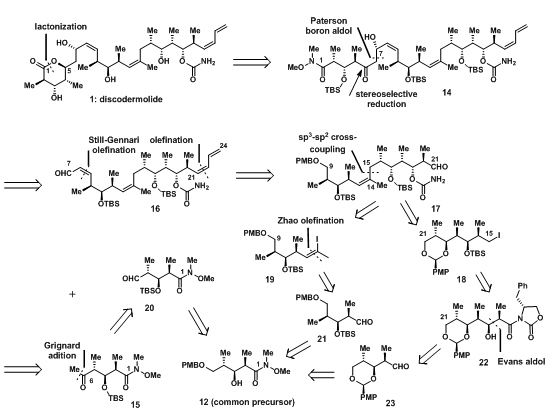
图3:Retrosynthetic Analysis of Discodermolide
2. Discodermolide的全合成
1). 共同前体12的合成
由于和Smith组一样选用weirneb酰胺作为共同前体,Mickel组对12的合成与Smith并无太大差异。但为了大量合成,他们对反应条件做了必要的改变。24是商业可得的原料,先上PMB基团保护羟基后,还原酯基为羟基,再用TEMPO氧化将其氧化成为成醛基,得27;27和28发生Evans Aldol反应,经过中间体29得到顺势产物30;再上weirneb酰胺中,原来的方法只能在THF中高效反应,但在放大反应中,THF会发生聚合,当制冷不好时,会有危险发生,故换用亲核试剂LiOOH,后还原中止得到不稳定的羧酸,再加入31使其形成羧酸盐,重结晶得到32,而原酰胺的另一半33也可通过重结晶回收;32在酸性条件下得到羧酸后,在发生两步反应得到共同前体12。
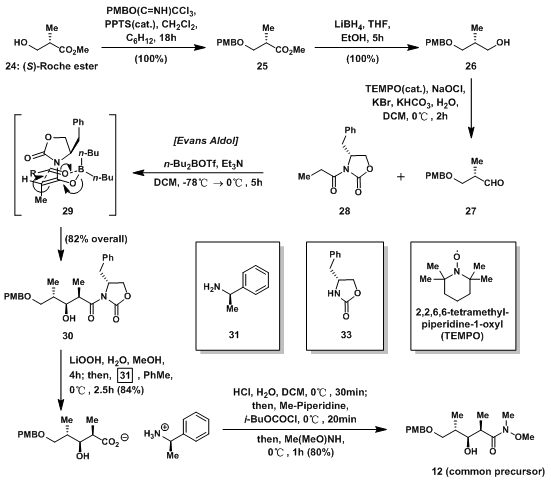
图4:Synthesis of Common Precursor 12
2). 3个碎片的合成
Mickel组的C1-C6碎片与Paterson组的C1-C6碎片有一定相似之处,区别在于一边为weirneb酰胺,另一个为酯基。12中的羟基先用TBS保护起来,后发生氢解反应,脱去PMB保护得到的羟基同样被TEMPO氧化成醛基20;20先与格氏试剂反应得二级醇,后经Parikh-Doering氧化得羰基,即碎片15。
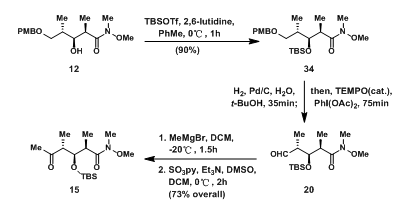
图5:Synthesis of C1-C6 Fragment 15
正如图6所示,碎片19的制备只需要两步,但是却遇到了许多麻烦。首先34被红铝还原成醛基,得益于weinreb酰胺的氧原子与Al原子配位形成稳定的四面体中间体35,使weinreb酰胺不会被还原成羟基;36先碘代生成37,37再与21反应,生成19。21与37反应时,Smith发现会有环氧化合物42的副产物生成。他们猜想,在两者反应时,有38与39两种中间体生成,38发生Zhao Olefination生成19,但39的O-P键瞬时断裂生成41所示结构,并且由于大基团相互作用,生成的氧负离子会发生Darzens-Type反应,离去I离子,生成环氧盐42,随后质子化生成43。虽然生成的19只有31%的收率,但是此反应可以大量反应,可以忍受其偏低产率。

图6:Synthesis of C9-C14 Fragment 19
在碎片18的合成中,12先经DDQ氧化,生成42(PMB羟基保护基),再将weinreb酰胺还原成醛基得23;随后发生Evans-Aldol反应得22后,先TBS保护羟基再将酰胺还原成羟基得45;45在羟基位碘代生成碎片18。
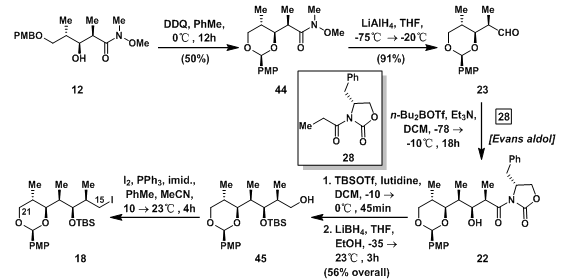
图7:Synthesis of C15-C21 Fragment 18
3). 碎片组合
碎片18发生Suzuki-Coupling生成48;48的羟基被氧化成醛基后,发生Nozaki-Hiyama-Kishi-Type Coupling反应,经50的过渡态得到52(major),再经Peterson Elimination得到53。之前第一步使用的是Negishi-Coupling,但发现其中会有副产物生成,故换用Suzuki-Coupling,另外,在另一个Nozaki-Hiyama-Kishi-Type Coupling反应中,受限于当时对此反应的认识,加入的是当量的镉试剂,有较大的浪费与污染。
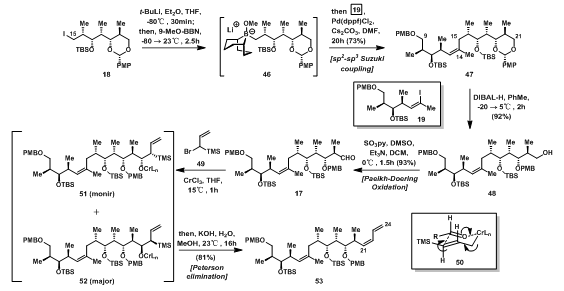
图8:Fragment Assembly and synthesis of triene 53
在16的合成中,先用DDQ脱去PMB保护基,后用TEMPO氧化,氧化端基上的伯醇得醛54,有趣的是,第二步反应中,加入催化量的水后,反应才能很好的进行;54先发生Wittig-Horner反应得56后,再与异氰酸酯57反应得到的58水解得到酰胺59,再将酯基还原成一级醇60;端基上的一级醇后又经TEMPO氧化得16。
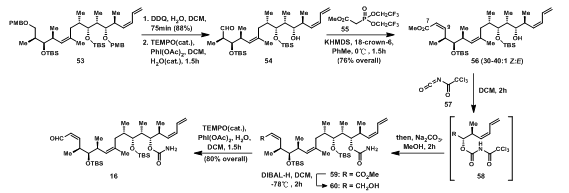
图9:Synthesis of α, β-Unsaturated Aldehyde 16
15与16发生Paterson Boron Aldol反应得到Discodermolide的结构骨架14和62。虽然只有一步,但Mickel曾抱怨到这是他遇到最难放大的实验,主要原因如下:首先,(+)-DIP-Cl容易吸潮且不稳定,其加入量就不好控制,于是采用商业上据称长期稳定的70%的己烷溶液;再者,产物14和62在用氧化性条件去除硼盐时不稳定,于是换用水来淬灭反应;最后,14和62两种产物的分离也是很大的问题,最终采用HPLC级纯的溶剂在反相色谱柱中得以分离,这当然会浪费大量的溶剂(20000L/60g产物),但最后几步了,浪费也没有办法了嘛。
需要说明的是,Paterson Boron Aldol反应因为在此处是甲基,立体控制与其他的大位阻基团有所不同,但是,立体控制,或其相应的过渡态并不为人们所认知。
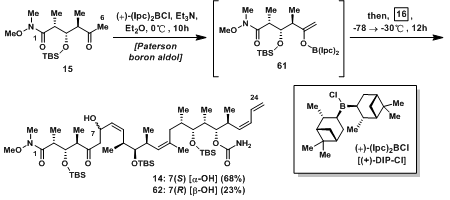
图10:Construction of the Discodermolide Carbon Skeleton
4). Discodermolide的全合成
首先,预期的aldol产物14的5位羰基被还原成羟基(Evans-Sakasena Reduction),然后在酸性条件下得到最终的产物Discodermolide。由于酸性条件下先形成环内酯,变成油状液体与水相分离,故需要用甲醇不断冲洗容器壁使反应能够进行完全。
当然,非预期的产物62分离时费了如此大的功夫,肯定得把它变成产物啦。同样的,先将5位的羰基还原成相应的羟基,再在酸性条件下形成环内酯67;7位的羟基可以先氧化在还原得到需要的手性结构,再于酸性条件下脱保护得到最终的产物Discodermolide。
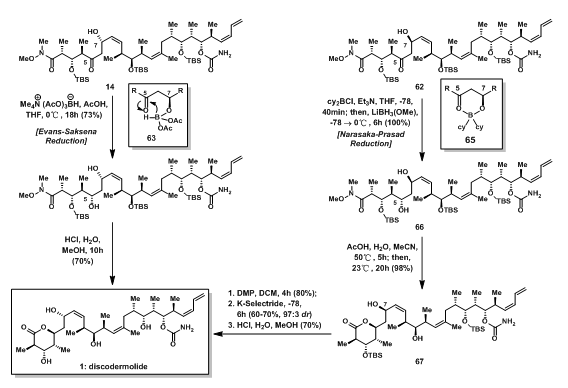
图11:Total Synthesis of Discodermolide
结语
这项工作介绍到这里就结束啦,所描述的问题只是其中的一部分,感兴趣的读者可以点击下面的文献链接查看原文[1]。正如Mickel所说:
“The success of this project and its chemistry paves the way for other, perhaps even more complex, naturalproducts to be prepared for early-phase clinical evaluations and sends a positive message to both isolation and synthetic academic community and possible other pharmaceutical companies that: ‘your work need not just be academic interest’ and it may be worth taking a few risks”.
可惜的是,2004年发现药物在临床上有巨大的副作用毒性,500g级的实验也被叫停了。但是KCN先生在后面说到:
“Although it didn’t lead to the manufacture of a drug, it set a standard for what is possible through total synthesis …… and the state of the art in organic synthesis continues to advance”.
有机合成就是如此的迷人,正如有机所的李昂老师所说,有的人生来就是做有机合成的,destined。那么下一篇就介绍命中注定的合成大师K. C. Nicolaou和D. A. Evans对Azaspiracid-1的全合成工作啦,希望大家喜欢。
参考文献
- a) Stuart J. Mickel, Gottfried H. Sedelmeier, Daniel Niederer, al. Org. Proc. Res. Dev., 2004, 8, 92; b) Stuart J. Mickel, Gottfried H. Sedelmeier, Daniel Niederer, et. al. Org. Proc. Res. Dev., 2004, 8, 101; c) Stuart J. Mickel, Gottfried H. Sedelmeier, Daniel Niederer, et. al. Org. Proc. Res. Dev., 2004, 8, 107; d) Stuart J. Mickel, Gottfried H. Sedelmeier, Daniel Niederer, et. al. Org. Proc. Res. Dev., 2004, 8, 113; e) Stuart J. Mickel, Gottfried H. Sedelmeier, Daniel Niederer, et. al. Org. Proc. Res. Dev., 2004, 8, 122;
- P. Gunasekera, M. Gunasekera, R. E. Longley, G. K. Schulte, J. Org. Chem., 1990, 55, 4912;
- B. SmithIII, Y-P. Qiu, D. R. Jones, K. Kobayashi, J. Am. Chem. Soc., 1995, 117, 12011;
- S. Harried, G. Yang, M.A. Strawn, D. C. Myles*, J. Org. Chem., 1997, 62, 6098;
- A. Marshall*, Z-H. Lu, B. A. Johns, J. Org. Chem., 1998, 63, 817;
- a) I. Paterson, G. J. Florence, K. Gerlach, J. P. Scott, Angew. Chem. Int. Ed. 2000, 39, 377; b) Ian Paterson*, G. J. Florence, K. Gerlach, J. P. Scott, N. Sereinig, J. Am. Chem. Soc., 2001, 123, 9535; c) I. Paterson*,I. Lyothier, J. Org. Chem., 2005, 70, 5494;
- Arefolov,J. S. Panek*, J. Am. Chem. Soc., 2005, 127, 5596;
- de Lemos, F-H. Poree, J-F. Betzer, A. Pancrazi, J. Ardisson, Angew. Chem. Int. Ed., 2007, 46, 1917;
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!



















No comments yet.