
作者:石油醚
引言
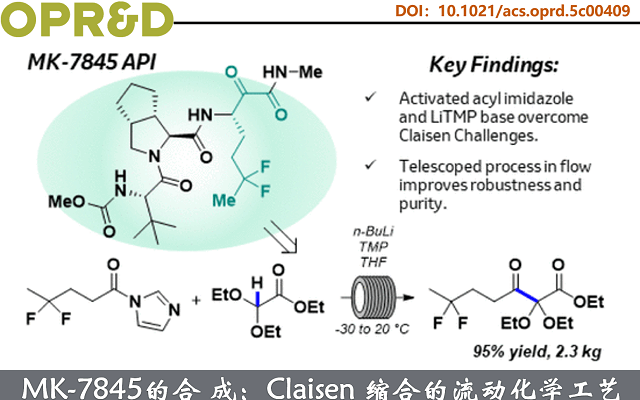
近日,Merck & Co., Inc.的科学家通过非典型交叉克莱森缩合开发了一条稳健的连续流合成路线,成功制备MK-7845关键中间体1,3-酮酯2。该工艺成功放大至2.3 kg规模,总收率达93%;所得酮酯2的THF溶液可直接用于下游工序,并高收率、高纯度地获得下一结晶中间体。
Development of a Claisen Condensation in Flow toward the Synthesis of MK-7845
Jeffrey S. Derrick,* Stasik Popov,* Cecilia Bottecchia, Ben W. H. Turnbull, François Lévesque,
Andrew J. Neel, Nastaran Salehi Marzijarani, Chibueze I. Onyeagusi, Douglas A. L. Otte,
Brittany Holden, Yining Ji, Alex Confer, Gilmar A. Brito, Yingju Xu, Michelle Zheng, Yu-hong Lam,
Jamie M. McCabe Dunn, and Mark Brower
Org. Process Res. Dev. 2026. Doi: 10.1021/acs.oprd.5c00395
正文
冠状病毒病(COVID-19)由SARS-CoV-2引起的一种高传播性传染病,2020年迅速演变为全球大流行,致死超700万人。尽管多种疫苗已获批使用,突破性感染仍时有发生,并可导致重症。然而,病毒变异频发、全球疫苗接种不均及保护效力下降,凸显了对安全有效抗病毒药物的迫切需求。其中,Meck的口服小分子药物MK-7845就是代表作。该分子含1,2-酮酰胺结构,其邻位手性中心极易差向异构化。逆合成分析表明,Ugi多组分反应是构建分子骨架的最优策略,可在晚期高效汇聚三个片段,中间体1为关键组分。
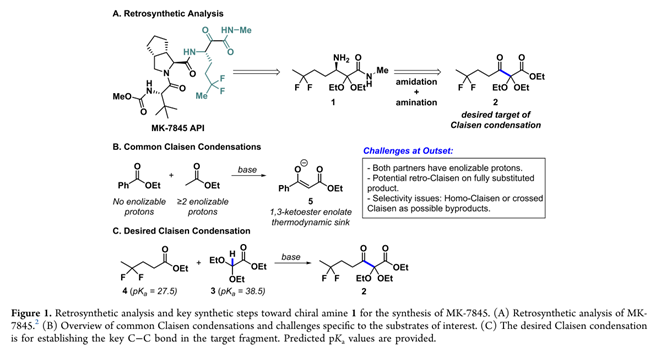
Merck & Co., Inc.的科学家利用β-酮酯2的连续不对称还原胺化/酰胺化路线(Figure 1A),来高效、低成本获得中间体1。其中,2可由廉价缩醛3与二氟酯4经交叉Claisen缩合制得(Figure 1C)。尽管该Claisen策略原子经济性高,但用于构建掩蔽型1,2,3-三羰基结构的报道极少,且从未用于API工艺开发。其主要原因在于底物反应性复杂(Figuren 1B):β-酮酯2无α-氢,无法形成稳定烯醇盐(如5),也易发生逆Claisen分解(缺乏热力学驱动力);而二氟酯4则易自烯醇化,导致副产物7及两类同缩合杂质。正因其显著的成本与效率优势,Meck的科学家着力突破上述反应性瓶颈,开发可行工艺。
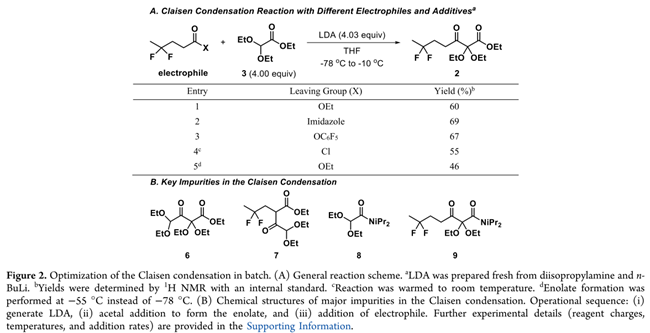
首先,作者对未活化乙酯4与亲核缩醛3在典型Claisen条件下的反应性进行研究。以LDA在−78 °C下脱质子化3生成烯醇盐,再与4反应,以60%产率得到目标产物2,但副产物较多(Figure 2A,Entry 1)。升温至−55 °C后产率降至46%(Entry 5),证实需严格低温控制。高碱用量(4.03 equiv)和低温可能源于烯醇盐中间体的固有不稳定,路易斯酸添加剂筛选未提升效率。因此,我们转向筛选活化亲电试剂替代4(Figure 2A)。在测试的四种试剂中,酰基咪唑10表现最优(Entry 2),产率较初始条件提高约10%,且易于制备,遂选定为后续工艺开发的亲电组分。
然而,使用10虽提升产率,反应纯度仍差:主要杂质为LDA加成产生的二异丙基酰胺8和9(Figure2B)。此外,还检出缩醛酯3自缩合生成的同Claisen产物6,以及不期望的交叉Claisen产物7。由于酮酯2常温下为油状,无法结晶纯化,亟需显著提升反应液纯度,以保障后续步骤中酮酰胺的顺利分离。

其次,作者使用空间位阻更大的锂二烷基胺替代LDA,来抑制杂质8和9的生成,我们筛选了。鉴于缩醛3的pKa较高(计算值38.5,Figure1C),优选碱的碱性应与LDA相近但位阻更大。此类碱由nBuLi在惰性气氛下缓慢滴加至相应胺中制备(避免放热及自燃风险)。LiTMP(Figure 3A,Entry 3)表现最优:以85%收率获得产物2,10 g规模结果可重现(Entry 8),且未检出TMP衍生的酰胺副产物(即类8/9结构)。其他醚类溶剂(Entries 4–5)未提升THF体系的产率。缩醛3用量可降至2 equiv(Entry 6),但进一步减少导致产率骤降至54%(Entry 7),归因于烯醇盐11易分解为酮烯12和LiOEt(Figure 3B)。鉴于其低温不稳定性及操作风险,转向连续流工艺——既可即时生成并消耗不稳定的烯醇盐11,又显著提升大规模生产的稳健性与可放大性。

基于上述研究,作者对连续流的合成工艺进行研究。
Claisen缩合的连续流工艺:工艺优化与反应器设计
采用分步策略尝试Claisen缩合:即先在流动中高效生成不稳定的烯醇盐11(精准控温、严格计量),再在线加入亲电试剂(Figure 4)。流出液转入间歇釜老化完成缩合。该法以LiTMP/酰基咪唑为体系,产率达95%,略高于小试间歇结果(Figure 3A, Entry 3),显著优于LDA体系(69%,Figure 2, Entry 2)。LiTMP与缩醛3用量可降至2.5或2 equiv(Entries 2–3),产率仍达93%和90%;进一步降至1.8或1.5 equiv(Entries 4–5)时产率微降,与间歇反应趋势一致(Figure 3, Entry 7)。若改用二氟乙酯4为亲电试剂,产率骤降至30%(Entry 6),证实LiTMP与酰基咪唑存在关键协同效应。所有使用酰基咪唑的流动反应均未检出TMP加合物或交叉克莱森副产物,凸显连续流在计量、时序与温度控制上的决定性优势。
鉴于LiTMP在THF中易分解,其质量直接影响工艺稳健性与放大可行性。新配LiTMP溶液若于室温放置数小时再使用,产率即降至42%(Entry 7)。虽低温储存(<−30 °C,THF:hexane中0.45 M)可延缓分解,但析出沉淀引发堵塞风险。为此,探索LiTMP的原位连续生成:由TMP与n-BuLi在流中即时制备,并立即与缩醛3反应消耗(Figure 5)。该策略既规避了大规模制备与深冷储存,又允许在更高温度下操作,彻底消除沉淀隐患。然而,三段式串联(LiTMP生成→烯醇化→缩合)存在工程复杂性。尝试两种简化方案:(1)前两步连续、缩合转间歇;(2)LiTMP与烯醇化连续耦合,酰基咪唑与烯醇盐11共进料至收集罐(无在线混合,。二者均未复现95%产率,证实反应对混合效率与计量精度高度敏感。最终确认:三阶段全连续流程(Figure 5)为最优解。
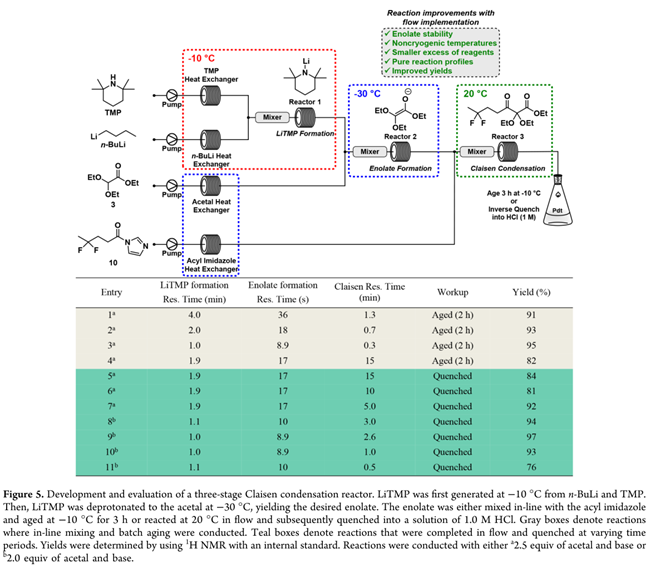
伸缩式三级连续流工艺的开发
多步连续流工艺的参数优化极具挑战:调整反应器体积以考察停留时间(τ)影响时,难以对各阶段τ进行独立系统筛选;且任一阶段的计量变动均会扰动后续反应。我们优先优化LiTMP生成的τ,并基于前期结果(Figure 4, Entry 2)设定烯醇化及与酰基咪唑10混合的τ。初始三段均设为−30 °C:LiTMP生成τ由1–4 min梯度考察(Figure 5 , Entries 1–3),产率波动微小,但同步检出新杂质(5–10%,源自未反应nBuLi对缩醛3的加成,即1,1-二乙氧基己烷-2-酮);即使TMP过量5%,该杂质仍无法抑制。将LiTMP生成温度升至−10 °C后,nBuLi完全消耗,杂质消失,总产率不受影响。
继而探索克莱森缩合全流程连续化——取消在线混合+间歇老化,改用平推流反应器直接完成缩合,以缩短周期、利于放大。升温至20 °C加速反应:τ=15 min时产率84%(Entry 4);后续于−10 °C再老化1 h无提升(Entry 5),证实15 min内反应已达终点。遂进一步缩短τ(Entries 6–11):仅当τ<1 min时产率明显下降(Entry 11, 76%)。最终确立最优三段序列:LiTMP生成(−10 °C, 1 min)→ 烯醇化(−30 °C, 8 s)→ 克莱森缩合(20 °C, 1 min),总产率93%(Entry 10)。反应液经出口直接加入1.0 M HCl溶液终止。
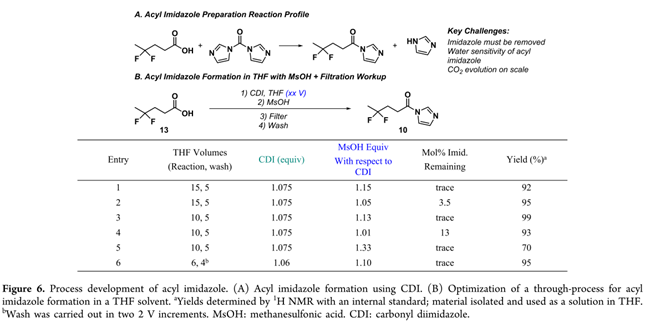
酰基咪唑的合成工艺开发
为支撑克莱森缩合全流程连续化,开发了酰基咪唑10的简化合成路线:CDI与二氟酸13在THF中反应后,直接加入等摩尔MsOH,将副产咪唑(pKa≈6.8)定量转化为低溶解度、高结晶性的咪唑鎓甲磺酸盐(pKa<5),经简单过滤即得高纯度酰基咪唑10溶液(92%收率,Figure 6B, Entry 1)。该方案规避传统水洗(Figure 6A),因酰基咪唑10遇水分解且THF与水互溶(Figure 6B)而不可行。所得溶液可直通下游反应,无性能损失。工艺稳健性研究表明:MsOH用量在0.95–1.05 equiv范围内(以咪唑计)对克莱森缩合产率与纯度无影响(SI, Section 3)。最终,反应与洗涤溶剂用量由15 V THF优化至10 V(Entry 6)。
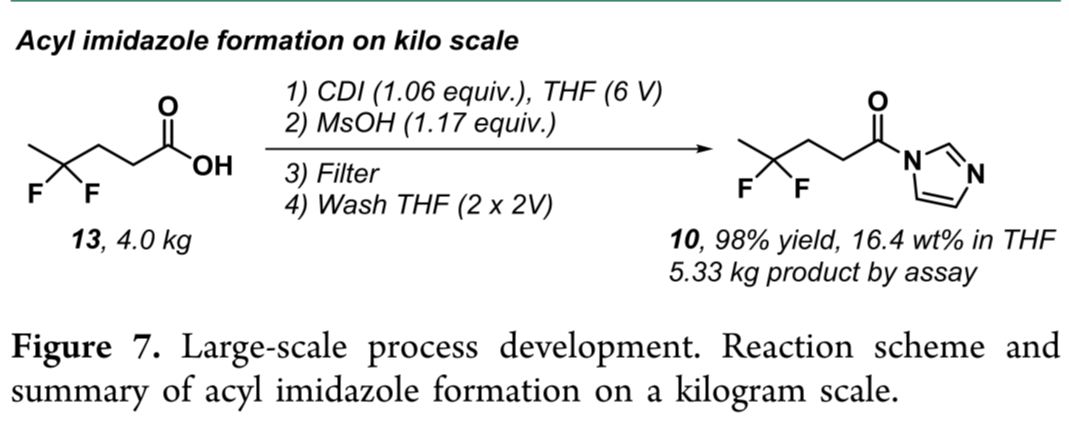
克莱森缩合反应的公斤级放大
综上,作者成功实现千克级工艺放大。放大前实施两项关键安全改进:(1)酰基咪唑合成中,于反应釜加装排气阀,并将酸13缓慢滴加入CDI悬浮液,以控制CO₂释放;(2)MsOH加入前预冷反应液至15 °C,规避放热风险。结合稳定性与过滤研究,4.0 kg规模下获得98%收率的酰基咪唑10 THF溶液(残留咪唑<2%,Figure 7),浓度16.4 wt%,与实验室批次一致。该溶液直通三阶段流反应,无需额外处理。
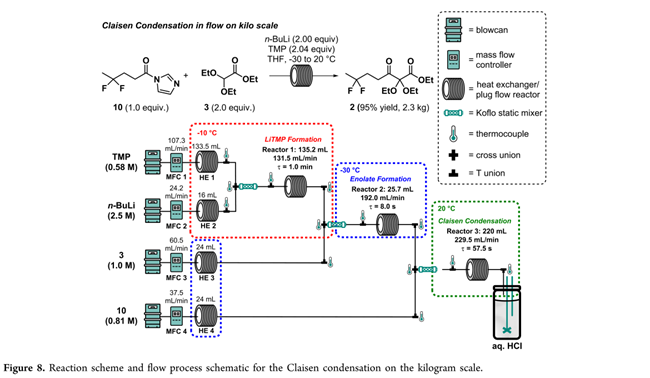
放大反应器设计(Figure 8)含两项核心升级:(1)采用氮气压力驱动替代机械泵,消除强碱对运动部件的腐蚀风险;(2)各试剂入口配置定制滤芯,并集成THF/甲醇吹扫罐与阀控汇流管,支持不拆卸快速冲洗。系统配备4个在线压力传感器(各进料流静态混合器上游)及多点热电偶实时监控。千克级运行中,nBuLi/TMP混合点出现瞬时压力波动,但未中断流动即自行恢复;事后确认为反应器1静态混合器入口Swagelok接头松动所致,已即时修复。温度偏差仅伴随压力波动发生,其余时段各阶段温度均精准稳定。最终,三阶段连续流工艺稳定运行4.5 h,产出酮酯2共2.3 kg,总收率93%,时空产率0.5 kg·h⁻¹。
结论
综上,Merck & Co., Inc.的科学家通过非典型交叉克莱森缩合开发了一条稳健的合成路线,成功制备MK-7845关键中间体1,3-酮酯2。作者进行了三项关键优化:(1)采用活化酰基咪唑酯作为亲电组分,显著提升反应选择性;(2)选用LiTMP为碱,高效生成烯醇化物,提高产率并简化后续纯化;(3)将整个转化设计为三阶段连续流过程,在THF中同步解决LiTMP溶解性、烯醇化物稳定性及反应参数精确控制(如混合计量、停留时间、温度切换)等难题,避免了传统批次操作所需的严格低温条件。该工艺成功放大至2.3 kg规模,总收率达93%;所得酮酯2的THF溶液可直接用于下游工序,并高收率、高纯度地获得下一结晶中间体。















No comments yet.