
作者:杉杉
导读:
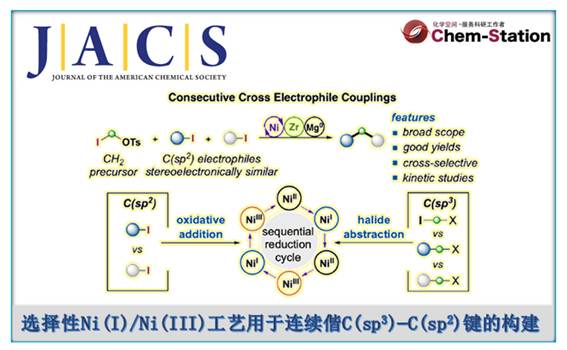
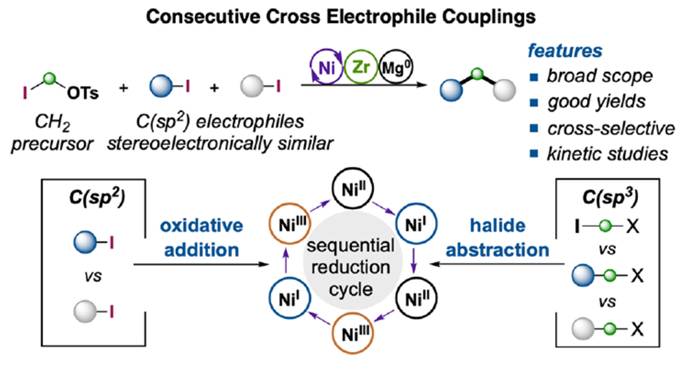
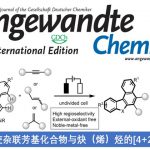
近日,上海科技大学的叶柏华课题组在J. Am. Chem. Soc.中发表论文,开发一种全新的连续开壳还原镍催化体系,促进了两种立体电性(stereoelectronically)相似C(sp2)-I反应物与亚甲基亲电试剂的反应,实现了偕C(sp3)−C(sp2)键的构建。其中,该策略以zirconaaziridine与元素Mg0作为还原剂,具有广泛的底物范围以及适用性。动力学研究表明,在镍催化中,zirconaaziridine-介导的氧化还原-转金属化过程促进了双重“串联还原”催化过程。值得注意的是,Ni(I)-I在C(sp2)-I键上的协同氧化加成,以及通过开壳C(sp2)-Ni(I)配合物在各种C(sp3)亲电试剂进行卤素原子攫取,均具有高度的选择选择性。
Selective Ni(I)/Ni(III) Process for Consecutive Geminal C(sp3)−C(sp2) Bond Formation
X. Li, Y. Gan, Y. Wang, B. Ye, J. Am. Chem. Soc. 2024, ASAP. doi: 10.1021/jacs.4c12581.
正文:
过渡金属催化的多组分反应在原子与步骤经济方面具有显著优势,能够在单反应操作中从多种起始底物高效构建复杂的分子骨架。然而,由于这些催化体系固有的复杂性,通常涉及两个或多个催化循环的过程,可能会导致严重的副反应,从而限制了反应的应用。目前,化学家们已经成功建立了多种过渡金属、电化学或光氧化还原催化剂促进烯烃的双官能化反应[1](Scheme 1A)。同时,现有的方法通常依赖于金属卡宾化学,利用重氮酯作为有效的单碳替代品。
镍催化的还原性交叉亲电偶联(XEC)反应已成为一种从有机亲电试剂中构建碳-碳键的强大合成方法。2014年,Weix课题组开发了一种镍催化芳基/烷基溴的XEC,在Fe(CO)5作为羰基替代物的存在下,以交叉选择性的方式合成了一系列非对称酮[2]。同时,使用氯甲酸酯作为CO前体也是有效的[3],通过单一操作成功合成了相应的烷基-烷基、芳基-烷基酮与C-酰基糖苷(Scheme 1B)。然而,在亚甲基CH2单元上连续引入两个有机单元的双重XEC工艺,却较少有相关的研究报道。传统上,构建此类骨架常依赖于Suzuki-Miyaura反应,要求在催化之前逐步制备相应的有机卤化物与硼酸酯[4](Scheme 1C)。作为对其他经典交叉偶联方法的补充,还建立了镍催化苯甲醚与苄基镁试剂的Kumada反应[5]。另一方面,使用苄基与芳香族亲电试剂进行的还原性镍催化提供了一种优越的解决方案,避免了转金属化偶联试剂的制备[6]。值得注意的是,还原剂的选择将显著影响催化途径。除了这些策略外,还探索了基于碳-氢或碳-杂原子键的直接官能化的方法[7],尽管位点选择性仍然存在挑战。前期,Zirconaziridine作为一种有效的氧化还原转金属化有机金属试剂,能够实现钯催化的(杂)芳香卤的交叉亲电偶联反应[8]以及镍催化非对映选择性C(sp2)糖基化反应[51]。受此启发,这里,上海科技大学的叶柏华课题组报道一种全新的连续开壳还原镍催化体系,促进了两种立体电性相似的C(sp2)-I反应物与亚甲基亲电试剂的反应,实现了偕C(sp3)−C(sp2)键的构建(Scheme 1D)。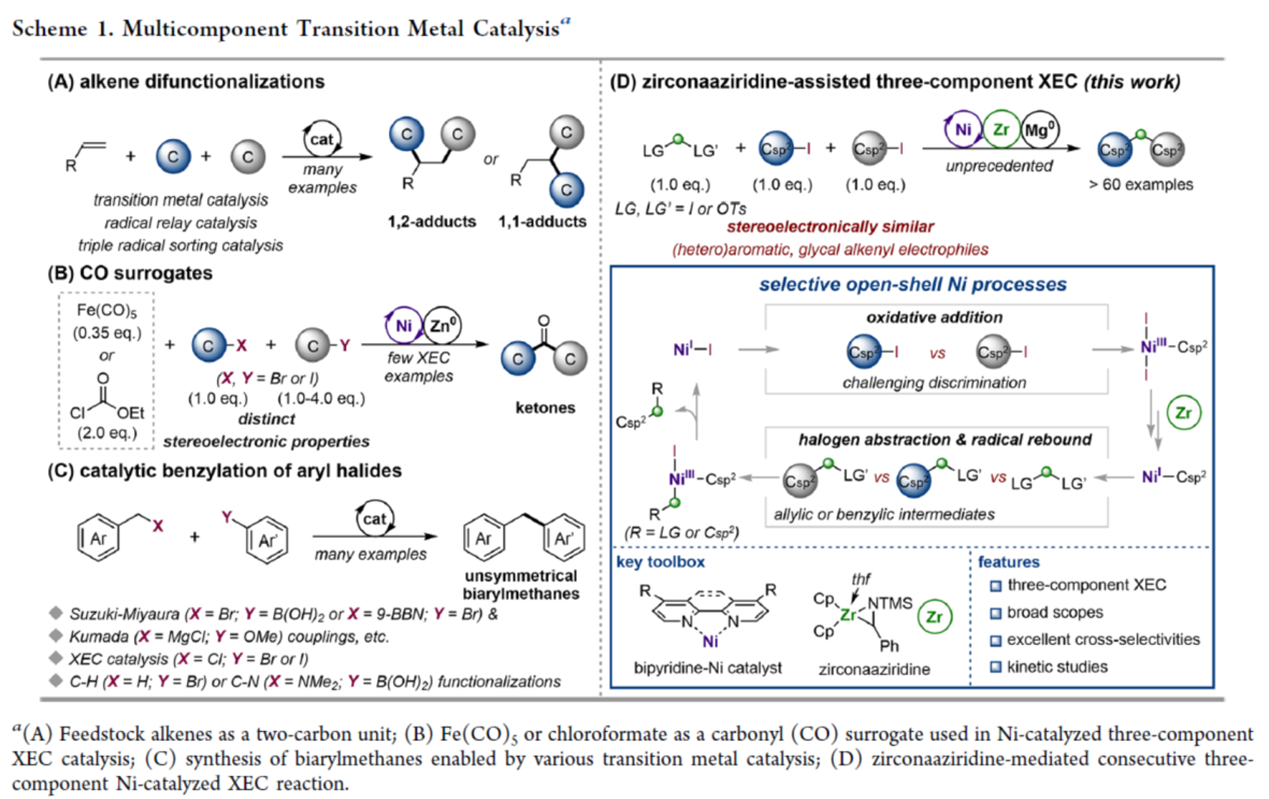
首先,作者采用葡萄糖碘化物(1a)与对碘异丙醚(3a)作为模型底物,进行相关反应条件的优化筛选 (Scheme 2)。进而确定最佳的反应条件为:采用2b作为亚甲基亲电试剂,L2-NiBr2作为催化剂,[Zr]与Mg0作为还原剂,在THF反应溶剂中,反应温度为25 oC,最终获得77%收率的产物4,交叉选择性为95%。其中,Mg0的引入导致联芳基homo-3a的增加以及痕量的homo-INT-1a。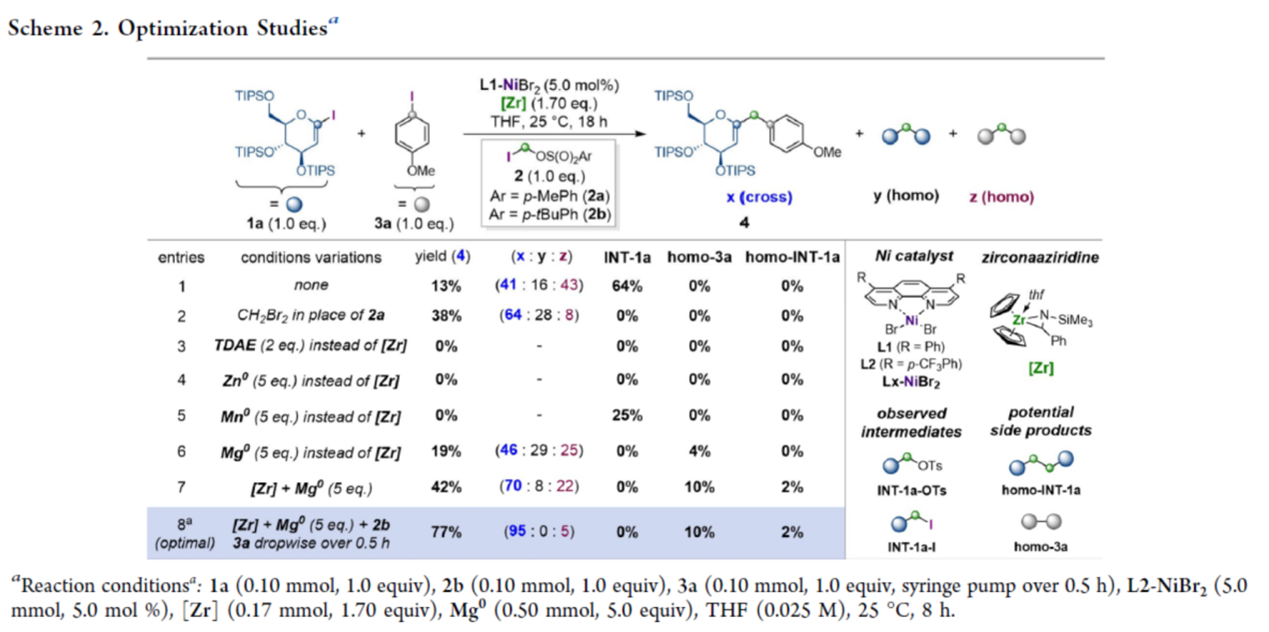
在上述的最佳反应条件下,作者对一系列亲电试剂的底物应用范围进行深入研究(Scheme 3)。首先,一系列不同电性取代的芳基与杂芳基碘,均可与1a与2b顺利进行反应,获得相应的产物4–26,收率为38-77%,交叉选择性为82-100% (Scheme 3A)。其次,各种烯基溴亲电试剂,也能够顺利进行反应,获得相应的产物27–34,收率为45-76%,交叉选择性为89-100% (Scheme 3B)。值得注意的是,烯丙醚(31–32)与胺(33)骨架在该条件下可保持完整。此外,该策略还可用于一些源自氨基酸、核苷与天然产物的医学相关亲电试剂的后期衍生化实验,获得相应的产物34–43,收率为39-75%,交叉选择性为82-100% (Scheme 3C)。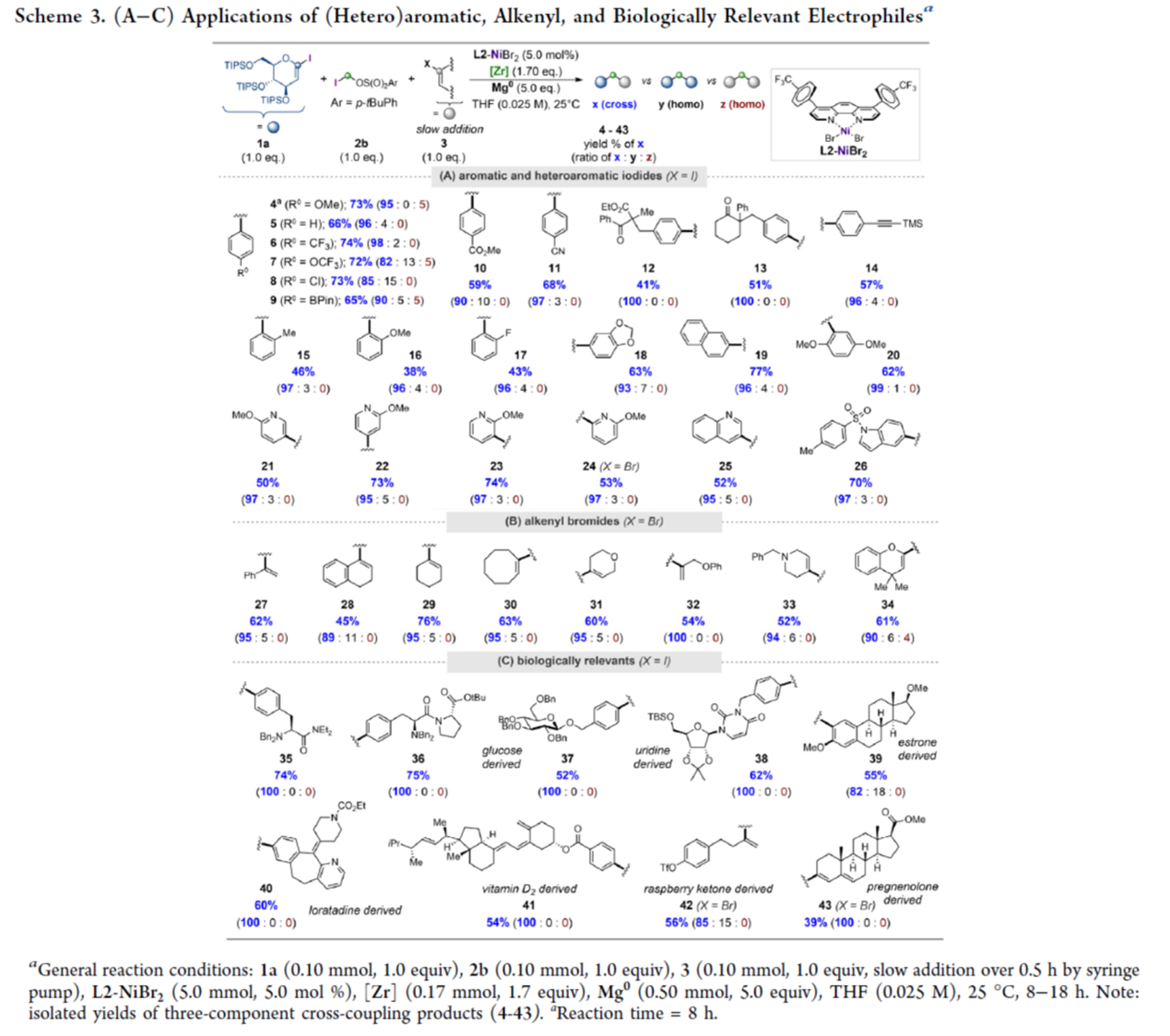
之后,作者发现,利用该策略,还可实现了非对称联芳基甲烷的合成,如44-55,收率为41-86%,交叉选择性为84-96%(Scheme 4A)。然而,在开发的反应条件下,用1,1-二溴戊烷代替标准的亚甲基亲电试剂2c仍然具有挑战性,只得到痕量的偶联产物。其次,该三组分XEC为经典烯丙基化提供了一种有价值的替代方案,可以合成各种烯丙基苯(56)与1-烯丙基烯糖(57–61)衍生物,收率为32-67%,交叉选择性为81-100%(Scheme 4B)。此外,该策略还可在生物活性分子上实现了苄基单元的后期引入(62–65)。在对C-糖基化化合物66进行后修饰后,可以很容易地获得潜在的SGLT2抑制剂C-葡萄糖苷69。值得注意的是,使用两种不同的糖基化碘化物(67)进行反应时,在硼氢化氧化过程后,可以97%的选择性形成C-二糖70。同时,通过对杂环化合物71(其是合成丙烯的潜在中间体)的逐步合成,进一步证明了反应的实用性。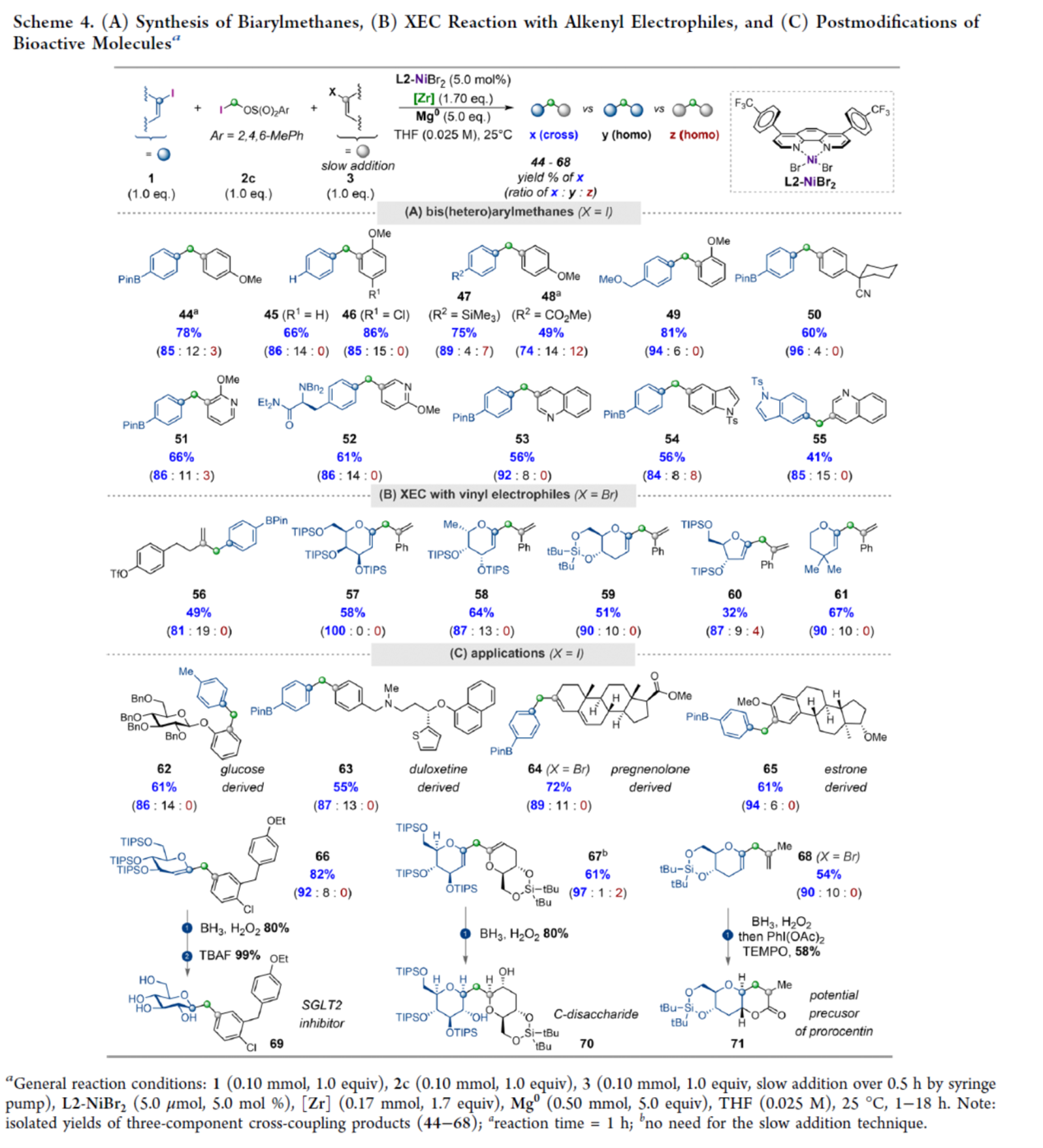
接下来,作者对三组分镍催化中交叉选择性的起源进行了研究 (Scheme 5)。首先,动力学研究发现,镍催化剂与C(sp3)亲电试剂呈正一级依赖性,表明它们在速率决定步骤中的关键作用,可能涉及用C(sp3)碘化物与低价Ni(I)配合物之间的卤原子攫取。同时,催化速率可能会受到C(sp2)亲电试剂选择的影响,即使它们不参与速率决定步骤(Scheme 5A)。其次,通过一系列竞争性实验进行Hammett plot研究表明,Ni(I)在C(sp2)-I键上进行协同氧化加成。更重要的是,zirconaaziridine在开壳镍催化中起着关键作用,能够选择性区分两种不同的C(sp2)-I亲电试剂(Scheme 5B)。反应速率取决于芳基碘亲电试剂的电子性质。一般来说,在芳烃上更大的电性差异导致两个芳基C(sp2)-I键之间的键离解能差异更大,从而导致更高的交叉选择性(Scheme 5C)。碘化聚糖1b的竞争性实验,形成的C(sp3)亲电中间体的反应性在随后的XEC循环中起着关键的作用,这也可能影响反应结果(Scheme 5D)。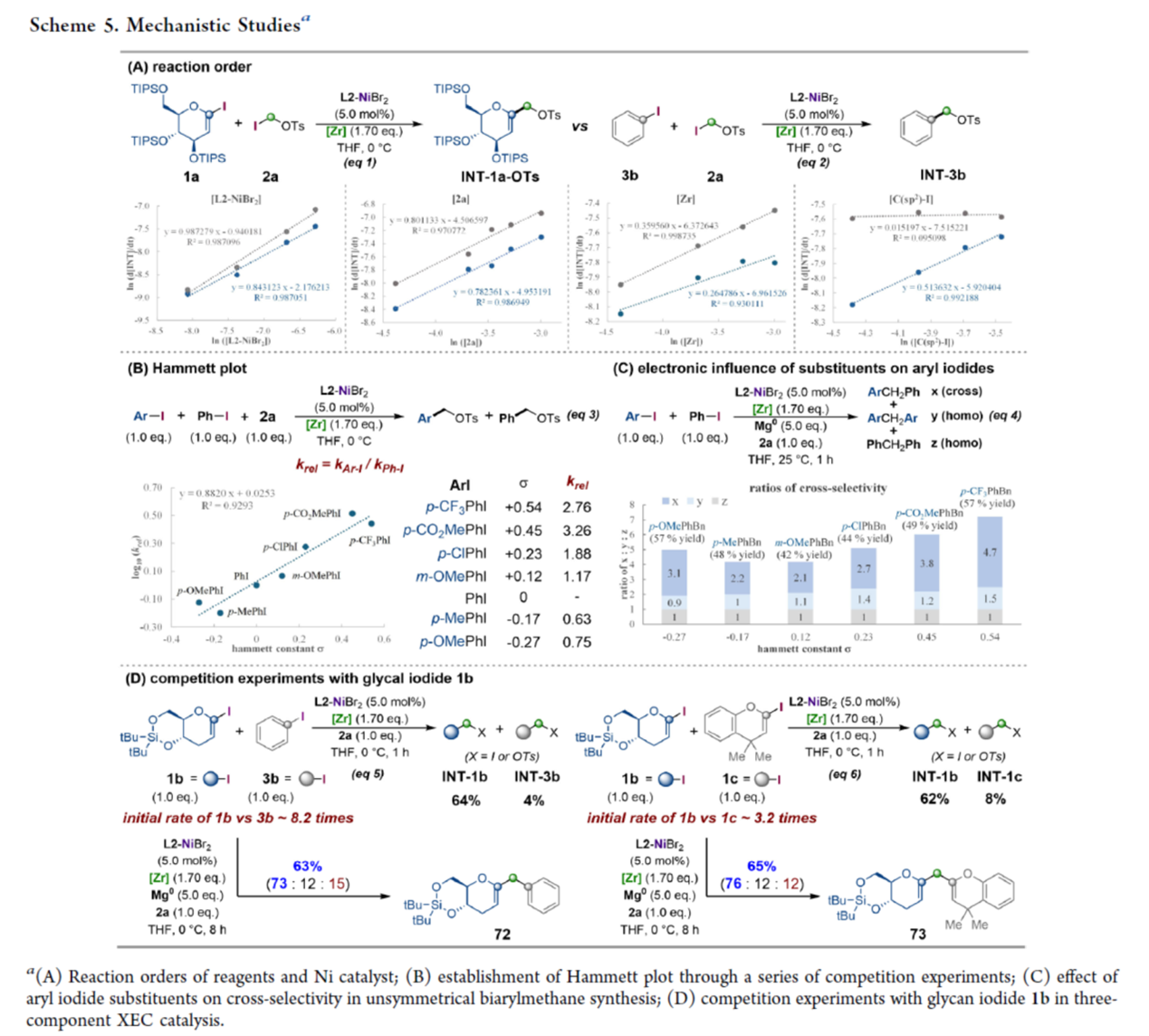
基于对上述C(sp2)碘化物反应性的机理见解,随后作者对涉及各种C(sp3)亲电试剂进行了一系列竞争性实验研究(Scheme 6A)。实验结果表明,zirconaaziridine-介导的XEC能够区分各种C(sp3)亲电试剂。其次,镍催化PhI与2a交叉偶联的初始反应速率比nhex-I快46倍(Scheme 6B)。反应性的显著差异可能归因于2a的C(sp3)-I键较弱,这在热力学上有利于通过低价Ni(I)配合物对碘化物进行攫取形成更稳定的C(sp3)自由基。因此,2a的快速初始XEC催化有效地抑制了不期望的自偶联。除了上述研究外,作者还进行了一系列对照实验来阐明Mg0的作用。Mg0可能促进了烯丙基或苄基碘中间体的单电子还原,生成相应的C(sp3)自由基中间体,在第二个XEC催化循环中重组为C(sp2)-Ni(II)中间体(Scheme 6C)。同时,卤盐可能作为添加剂,可以加速Ni(II)的还原过程。
基于上述的实验研究,作者提出如下合理的反应机理 (Scheme 6D)。在第一个催化循环中,Ni(I)-I配合物A氧化加成到两种C(sp2)碘化物中的一种,其中观察到的选择性可归因于C(sp2)-碘化物之间的键强度差异及其初始浓度的差异。接下来,用Cp2Zr(III)还原所得的C(sp2)-Ni(III)配合物B将生成C(sp2)-Ni(II)中间体C。随后,中间体C通过氧化还原转金属化过程,可将其化为低价双金属Ni-Zr配合物D。然后,这种关键的开壳Ni(I)配合物可能会通过设想的卤原子攫取与亚甲基亲电试剂2反应,生成自由基结合的Ni(II)中间体E。最后,在自由基重组后,形成的C(sp2)-Ni(III)-C(sp3)配合物F。配合物F经还原消除,生成C(sp2)-CH2-OTs中间体。在串联的XEC循环中,Mg0可能作为强效单电子还原剂。在碳自由基重组为C(sp2)-Ni(II)配合物H后,随后还原消除Ni(III)配合物J从而获得所需的交叉偶联产物,同时再生了Ni(I)-I配合物A。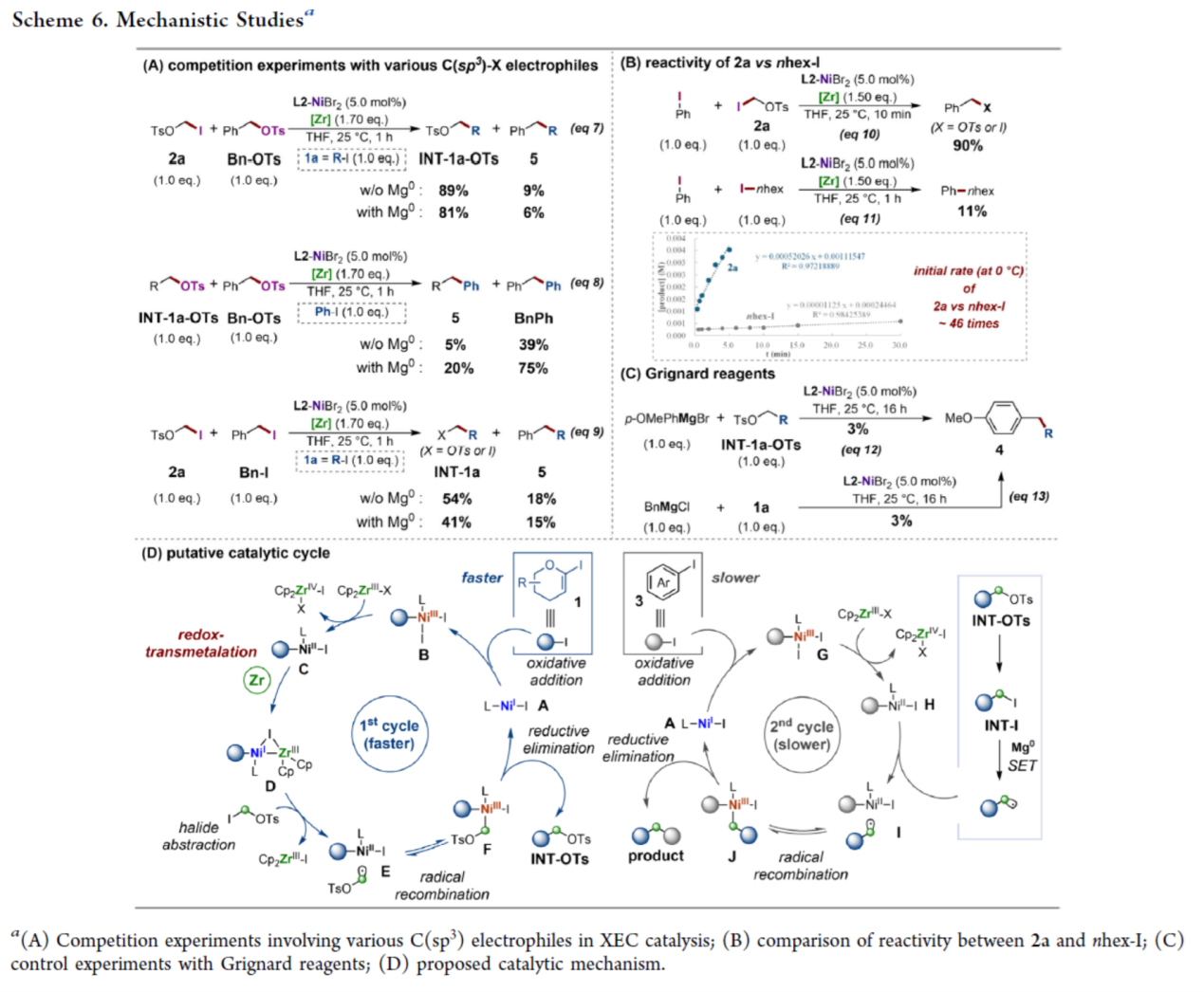
总结:
上海科技大学的叶柏华课题组报道一种由选择性开壳镍催化体系促进的三组分XEC反应。在这种方法中,zirconaaziridine充当一种强大的氧化还原转金属化试剂,产生低价C(sp2)-Ni(I)配合物,促进在速率决定步骤中选择性区分各种C(sp3)亲电试剂。此外,在Ni(I)-I协同氧化加成过程中区分两种不同的C(sp2)-碘化物的能力对于实现高交叉选择性至关重要。
参考文献:
[1] J. Z. Wang, W. L. Lyon, D. W. C. MacMillan, Nature 2024, 628, 104. doi:
[2] A. C. Wotal, R. D. Ribson, D. J. Weix, Organometallics 2014, 33, 5874. doi:
[3] R. Shi, X. Hu, Angew. Chem., Int. Ed. 2019, 58, 7454. doi:10.1002/anie.201903330.
[4] M. Nambo, E. C. Keske, J. P. G. Rygus, J. C. H. Yim, C. M. Crudden, ACS Catal. 2017, 7, 1108. doi:10.1021/acscatal.6b03434.
[5] M. Tobisu, T. Takahira, N. Chatani, Org. Lett. 2015, 17, 4352. doi:10.1021/acs.orglett.5b02200.
[6] J. Zhang, G. Lu, J. Xu, H. Sun, Q. Shen, Org. Lett. 2016, 18, 2860. doi:10.1021/acs.orglett.6b01134.
[7] P. R. Ledwith, M. L. Cooney, K. A. Bahou, J. Garcia-Carceles, J. Thomson, J. Bower, Angew. Chem., Int. Ed. 2024, 63, No. e202411555. doi:10.1002/anie.202411555.
[8] T. Wu, Y. Zhang, Y. Fu, F. Liu, J. Tang, P. Liu, F. D. Toste, B. Ye, Chem 2021, 7, 1963. doi:10.1016/j.chempr.2021.06.007.
[9] Y. Gan, J. Zhou, X. Li, J. Liu, F. Liu, X. Hong, B. Ye, J. Am. Chem. Soc. 2024, 146, 16753. doi:10.1021/jacs.4c04587.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
关注Chem-Station抖音号:79473891841
请登陆TCI试剂官网查看更多内容
https://www.tcichemicals.com/CN/zh/














No comments yet.