
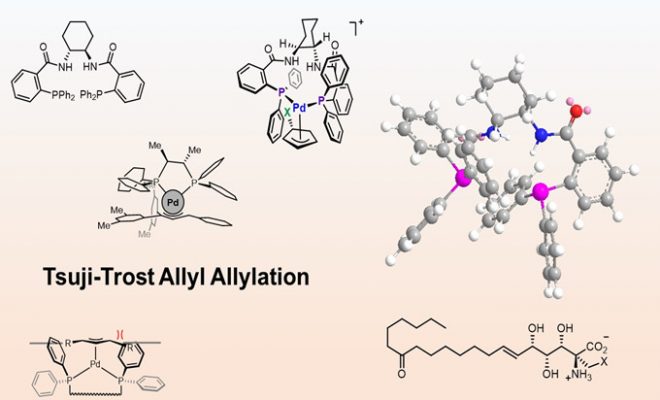
本文作者:孙苏赟
第一部分 基本原理和烯丙基异构化
在之前,我详细的介绍了不对称羰基化合物及其衍生物的烯丙基化反应及其一些应用,而Tsuji-Trost烯丙基取代反应是合成中用于烯丙基化合物衍生化的重要方法之一。Tsuji-Trost反应是烯丙基化学的重要代表,自从其被发现以来已开发出多种可用于反应的过渡金属和亲核试剂。其中,亲核试剂的取代可以发生在端位碳和取代基近端上,但是在多数情况下是以端位碳发生反应。从机理上看反应的结果和反应中间体σ-/π-烯丙基金属配合物之间的平衡和性质有关。
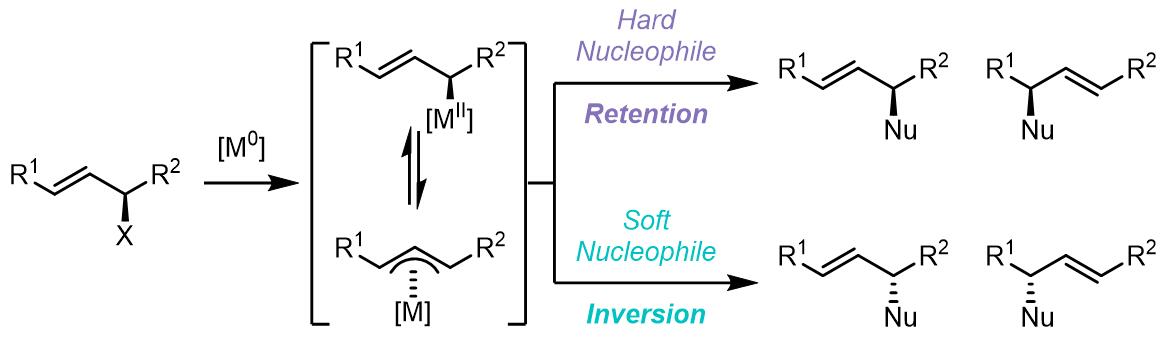
1. 反应机理
这个过程和Wacker氧化的原理相似,过渡金属和烯丙基共轭体系配合使其亲电性增加而可以被亲核试剂进攻。反应从低价金属和双键的氧化加成开始,形成一个烯丙基配合物,得到高价太过渡金属。根据配体和烯丙基的离去基团的不同,或配合物自身的性质,配合物中金属中心可以是电中性的,也可以阳离子。多数情况下,反应的亲核试剂是较软的碳负离子,且常常在位阻更小的端位碳碳上进行取代,生成的双键-金属配合物又发生解离得到产物并且使得催化剂重生。

反应中亲核试剂的取代步骤一般(但不绝对是)是不可逆的,同时也有一些亲核取代后的中间体发生异构化的例子。反应的整个过程都与金属种类和配体高度相关,并且反应也可以被添加剂影响。
2. 立体化学
从底物的立体化学上来看,有以下几种情况:
(1) 内消旋化合物作为底物时,产物的手性是在氧化和原步骤引入的。例如:
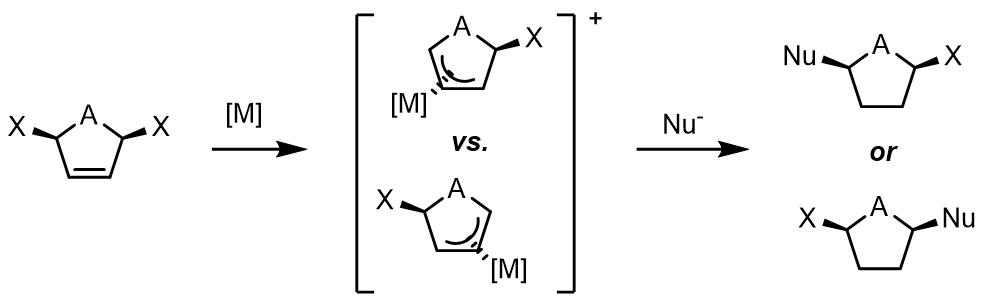
(2) 对于1,3-位置的取代基是相同的烯丙基化合物,如果金属配合物没有手性,那么反应中间体的烯丙基化合物没有手性,且亲核试剂进攻没有面选择性;反之如果金属配体由手性则可以产生反应的面选择性。
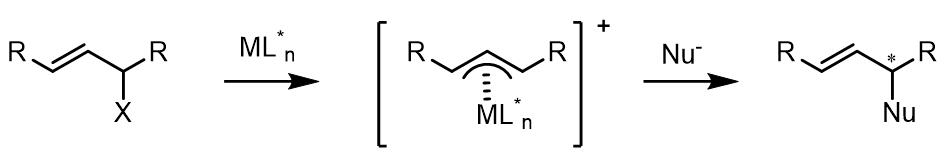
(3) 1,1-位置取代基是相同的化合物和外消旋烯丙基化合物在Tsuji-Trost反应中是潜手性中心。对于潜手性化合物,反应的立体化学选择性发生在亲核试剂的取代步骤:
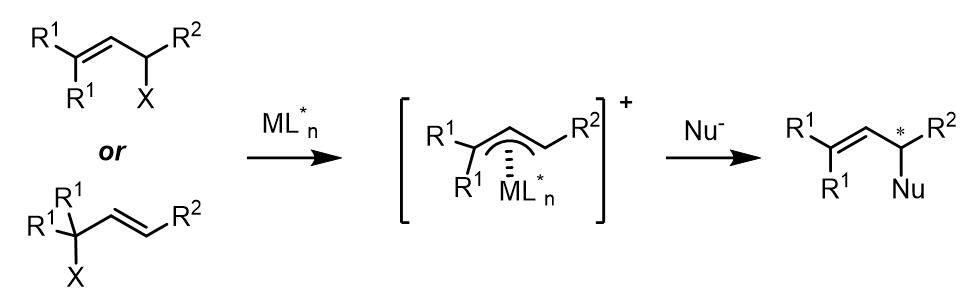
(4) 对于反应中使用潜手性的亲核试剂,如果金属配体中是非手性配体,那么反应的立体选择性则不会很好:
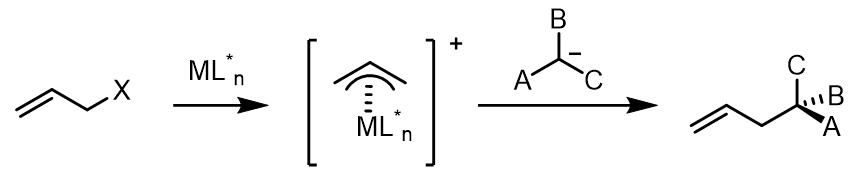
(5) 如若在金属配合物中使用配体使得烯丙基金属配合物不能发生构型翻转,那么产物的对映异构体比例则不会有变化。因此在此类反应在可以进行动力学拆分反应。
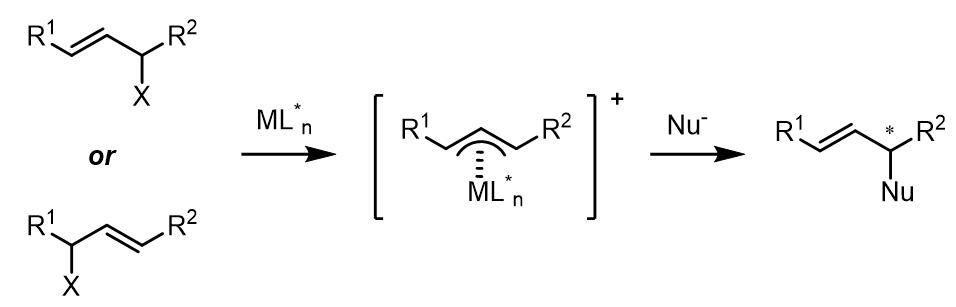
3. 亲核试剂进攻的区域选择性
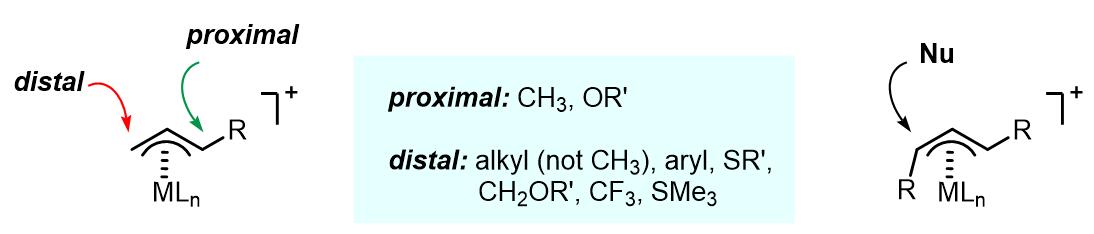
亲核试剂进攻的区域选择性和反应中的金属有很大的关系,以Pd为例,一般有以下几个影响因素:
(1) 烯丙基上的位阻因素和电子密度。亲核试剂更加倾向于进攻烯丙基上位阻更小的碳原子。但是位阻因素的影响不如电子密度的影响大。例如1-甲基和1,1-二甲基的烯丙基配合物使得亲核试剂的进攻倾向于发生在取代基近端碳原子上。
(2) 共轭取代基的影响。对于烯丙基上有例如苯基,羰基,氰基等取代基的底物,亲核试剂倾向于发生在端位,这样得到的配合物的稳定性更好。
(3) σ-吸电基团,例如CH2OR, CF3, 倾向于端位进攻。
(4) 在其他条件相同的情况下,RO–亲核试剂倾向于取代基近端进攻,RO–和SiMe3倾向于端位进攻。
(5) 配体的性质会影响过渡金属上的电子密度,因此影响亲核试剂的进攻位置[9]。
(6) 烯丙基上取代基和配体都会和亲核试剂发生相互作用,例如氢键作用。
值得注意的是,这些经验总结是基于动力学控制的反应的。热力学控制的反应不一定遵循以上的大规律。
4. 烯丙基配合物的异构化
烯丙基配合物的异构化可能有以下几种情况:
(1)取代基的syn-/anti-异构化
烯丙基金属配合物中间体是反应的关键所在,在配合物的异构化中,四种结构常常处在动态的平衡之中。烯丙基的构型一般以其上取代基与2-位的氢的相对位置而确定:
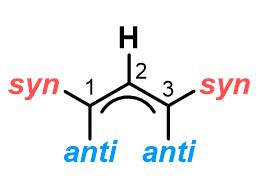
syn-和anti-取代基可以快速发生异构化而调换位置,即配合物A和D 可以通过π-σ-π过程进行异构化,这也是最重要的异构化步骤;σ-配合物B和C之间又可以发生构象的转变成,这也就是为什么在第(3)类反应中即使底物不相同但是得到的产物是相同的原因。其中,A和D之间是否可以进行直接转化还存有争议。

(2)烯丙基的表观旋转效应
目前尚未有证据表明烯丙基可以围绕烯丙基配体-Pd的连线作为轴旋转,但是目前提出了一个这种的说法,依旧经过π-σ-π过程,但是这个过程中不会发生syn-/anti-取代基的异构化,而是发生C-M键的旋转:
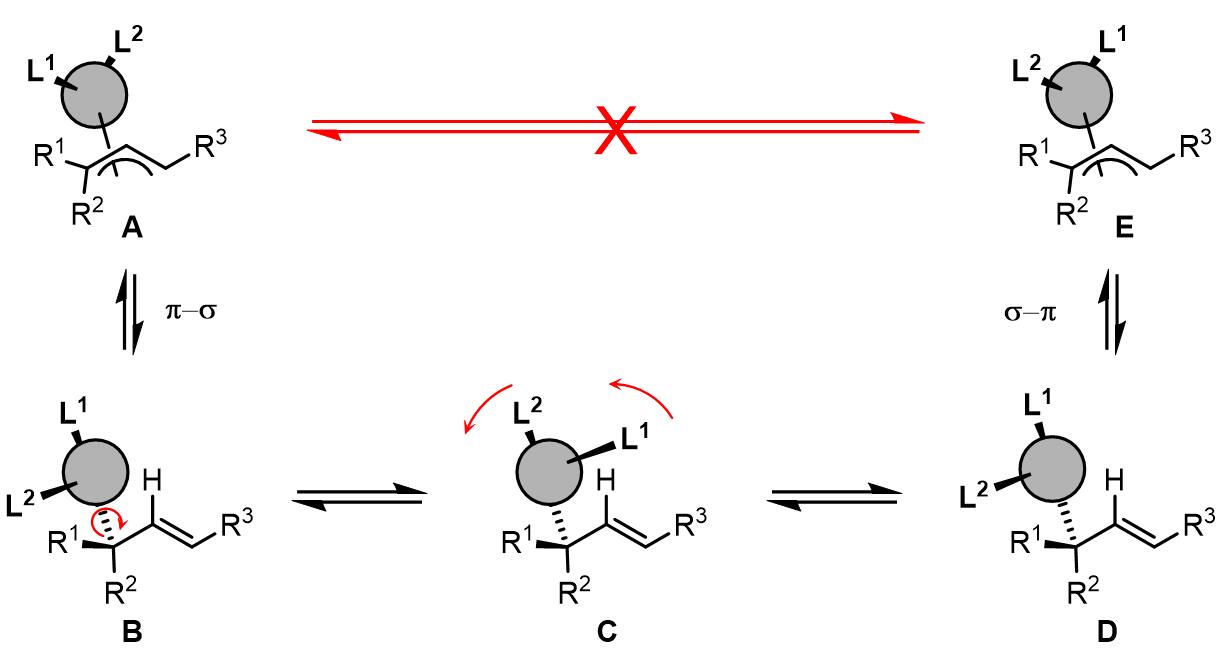
这个过程烯丙基取代基高度相关,普遍认为如果烯丙基取代基是强供电基团,配体是弱供电基团或吸电基团,那么对trans-PdLR2到cis– PdLR2是有利的。以下就是一个烯丙基旋转的例子[1]:
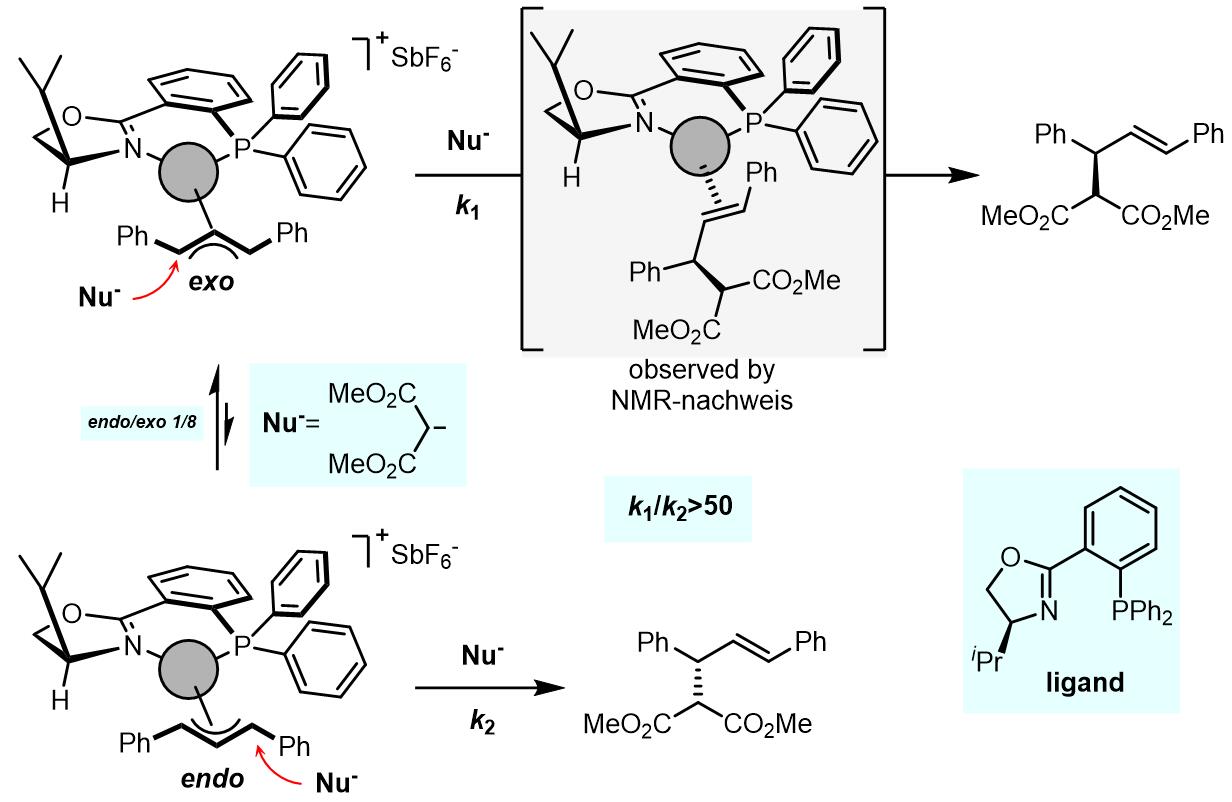
在相同的反应条件下, endo-配合物被亲核试剂进攻的速率至少要比exo-配合物的反应快50倍,但是exo-/endo-配合物的比例和溶剂组成高度相关。
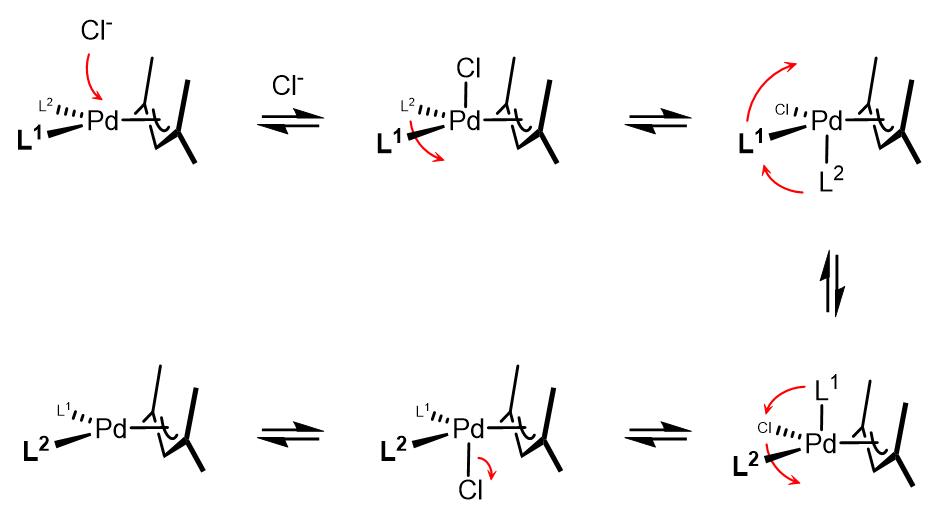
(3)烯丙基表观交换和假旋转
在反应体系中加入Cl–可以加速烯丙基的表观旋转现象,因此使用烯丙基氯化钯二聚体是一个很好的选择[2,3]。Åkermark和Vitagliano提出了烯丙基假旋转理论[4]:
(4)配体解离导致的异构化
最后一种可能的异构化过程是配体解离-重配合的机理:
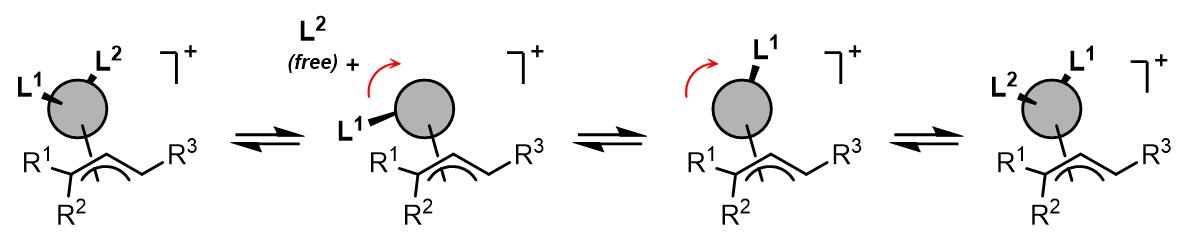
在这个过程中,金属上的一个配体会发生解离,此时另外一个类体发生构型翻转,最后解离的配体重新与过渡金属配合而完成金属构型的异构化过程。此类过程在双齿氮配体之中较为常见,例如2,2’-联嘧啶和TMEDA作为配体时聚会发生此过程(反应过程由EXSY谱图检测到)[5,6]:
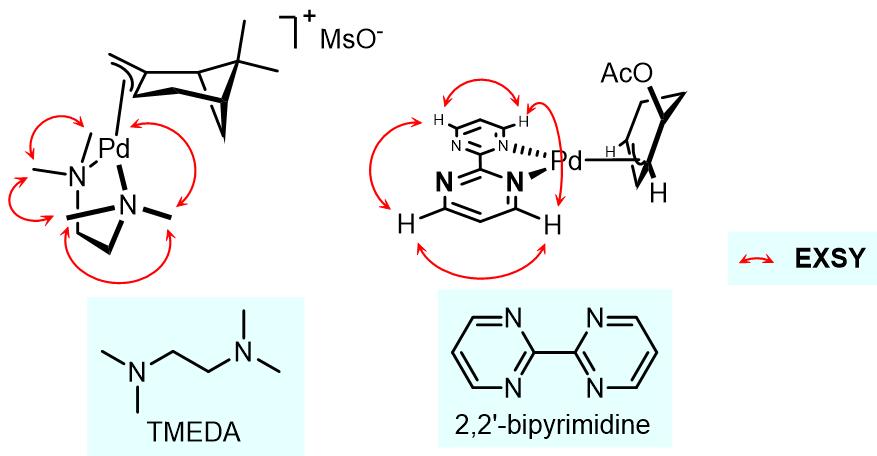
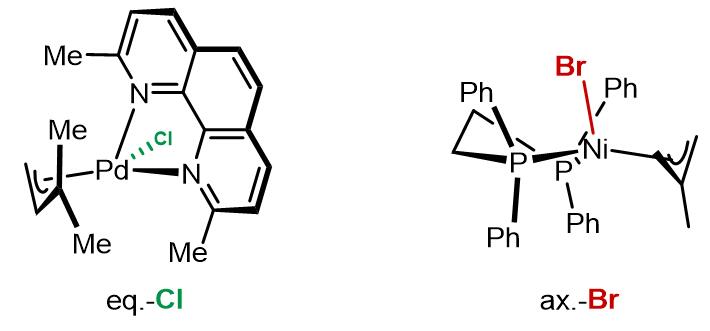
但是仍有争议的是这个过程是否是氯离子诱导的解离-重组过程。左边配合物的氯原子在eq.键上,右边配合物溴原子在ax.键上,导致这两个卤素原子在不同位置的因素不完全的是电子因素,也可能有一部分原因是因为位阻因素[7,8]。
References
- [1]Tetrahedron Lett., 35, 10, 1523 – 1526. DOI: 10.1016/S0040-4039(00)76748-7
- [2] Chem. Eur. J. 1995, 1, 12. 10.1002/chem.19950010106
- [3] J. Am. Chem. Soc. 1994, 116, 8, 3613–3614. DOI: 10.1021/ja00087a063
- [4] Organometallics 1993, 12, 12, 4940–4948. DOI: 10.1021/om00036a038
- [5] J. Am. Chem. Soc. 1994, 116, 8, 3631–3632. DOI: 10.1021/ja00087a072
- [6] Organometallics 1991, 10, 6, 1800–1806. DOI: 1021/om00052a028
- [7] Organometallics 1993, 12, 12, 4940–4948. DOI: 1021/om00036a038
- [8] J. Chem. Soc. A. 1970, 206 . DOI: doi.org/10.1039/J19700000203
- [9] Synthesis 2008(17): 2669-2679. DOI: 10.1055/s-2008-1067220
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!














No comments yet.