
作者:杉杉
导读:
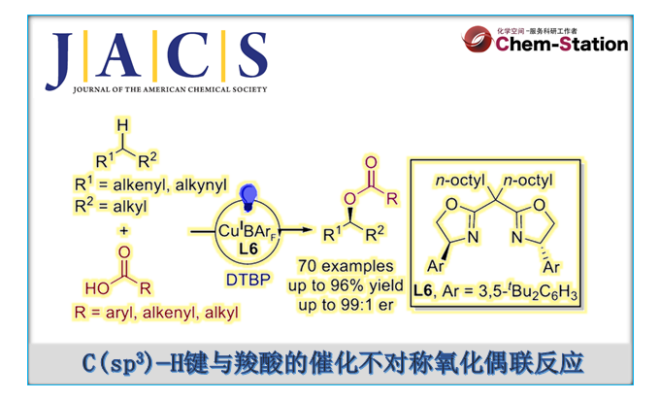
近日,南开大学的周其林团队在J. Am. Chem. Soc.中发表论文,报道一种全新的羧酸与C(sp3)−H键的高度对映选择性氧化偶联反应。其中,该方法适用于烯丙基与炔丙基C−H键,并以各种羧酸作为供氧试剂(oxygenating agents)。此外,该方法可从易得的烯烃与炔烃中直接合成了一系列手性酯,大大简化了手性酯与相关醇的合成。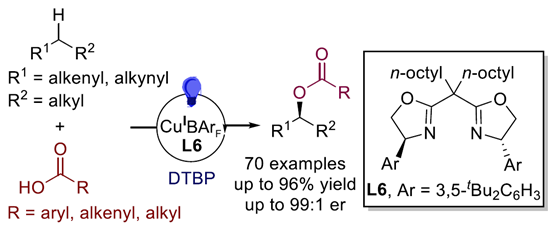
Catalytic Asymmetric Oxidative Coupling between C(sp3)−H Bonds and Carboxylic Acids
X. Liu, F. Li, T. Wang, L. Dai, Y. Yang, N. Jiang, L. Xue, J. Liu, X. Xue, L. Xiao, Q. Zhou,
J. Am. Chem. Soc. 2024, ASAP. doi: 10.1021/jacs.4c12544.
正文:
有机分子中C(sp3)−H键的直接对映选择性官能团化是一类极为重要的反应,具有显著的原子经济性与步骤经济性。其中,C(sp3)−H键的对映选择性氧化反应可直接将氧官能团引入烷烃中,在新药的发现与开发方面具有巨大的潜力。同时,手性含氧脂肪族骨架也广泛存在于各类天然产物与药物分子等中。最近,诸多研究团队已经成功开发大量过渡金属催化C(sp3)−H键对映选择性构建C−C与C−N键的反应[1]。然而,通过C(sp3)−H键氧化实现C−O键的对映选择性构建,仍然是一个巨大的挑战[2] (Figure 1A)。在自然界中,酶催化选择性C(sp3)−H键氧化方面表现出显著的效率。然而,开发用于C(sp3)−H键对映选择性氧化的分子催化剂需要克服C−H键固有的低反应性,并且需避免过度氧化生成酮。1958年,Kharasch团队开发了一种铜催化烯烃中烯丙基C(sp3)−H键氧化反应(即Kharasch反应),合成了一系列烯丙基酯[3] ,但仅限于环状烯烃与过氧化物试剂(Figure 1B)。受到前期铜催化苄基C−H键的不对称胺化反应[1]的启发,这里,南开大学的周其林团队报道一种全新的在蓝光照射下使用阳离子铜催化剂,实现了羧酸与C(sp3)−H键的高度对映选择性氧化偶联反应,克服了Kharasch反应的局限性,并具有广泛的底物范围以及优异的对映选择性等特点(Figure 1C)。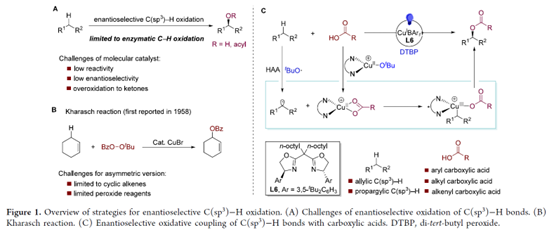
首先,作者采用(E)-1-苯基-1-丁烯(1a)与4-甲氧基苯甲酸(2a)作为模型底物,进行相关反应条件的优化筛选 (Table 1)。通过对反应条件的大量筛选(如各种铜源、配体、氧化剂、溶剂、温度与光源),进而确定最佳的反应条件为:采用CuCl作为催化剂,NaBArF作为添加剂,L6作为配体,DTBP作为氧化剂,C6F6作为溶剂,12 W 395 nm 蓝色 LED作为光源,最终获得82%分离收率的产物3a,E/Z为>20:1,er为96:4。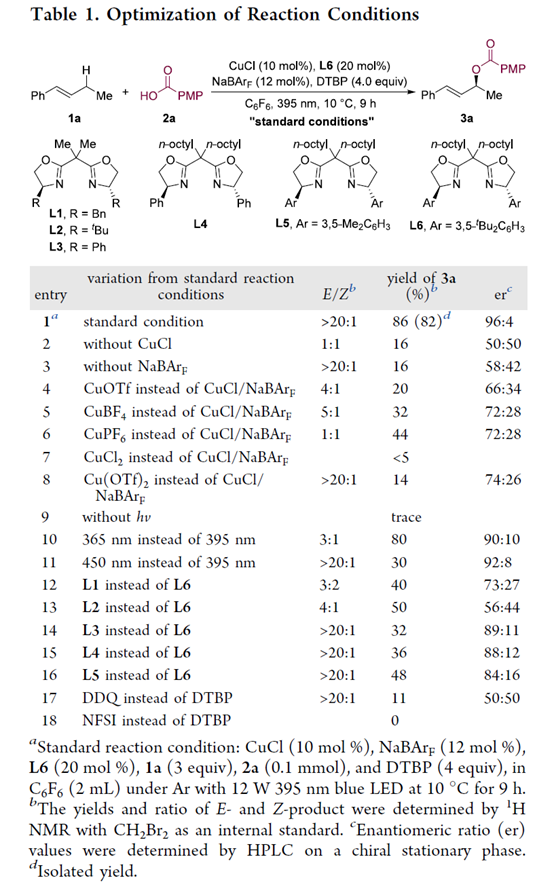
紧接着,作者对烯烃C(sp3)−H键与羧酸参与对映选择性氧化偶联反应的底物范围进行了扩展(Table 2)。首先,烯烃底物中苯环对位的取代基,对反应的对映选择性影响很小(3b–3k,er 为91:9-98:2),但强吸电子基团p-CF3(3e)导致产率降低(40%)。值得注意的是,在同时存在苄基与烯丙基C-H键的情况下,氧化仅发生在烯丙基C-H键(3h)。由于空间效应,苯环上含有间位(3l–3n与3q)与邻位取代基(3o与3p)的底物,具有更高的对映选择性(er 为94:6-99:1)。然而,3-溴-(3n,45%)与3,5-二氯取代(3q,38%)的底物,产率较低。含有2-萘环的烯烃底物也表现良好,可以51%的收率与93:7 er得到了所需的产物3r。同时,含有不同烷基链的底物,也具有良好的兼容性,得到了相应的手性烯丙基酯(3s–3w),收率为51-70%,er为92:8-98:2。其次,一系列不同取代的芳香酸与杂芳香酸,均可以良好的收率(69-88%)与高度的对映选择性(91:9-96:4 er)获得相应的产物3x–3ae。具有不同烷基链、螺环与酮基的脂肪酸,也可进行反应,具有出色的的对映选择性(3af–3am,94:6-96:4 er)。此外,不饱和肉桂酸还可以与烯烃1a反应,产生具有优异对映选择性(98:2 er)的氧化偶联产物3an。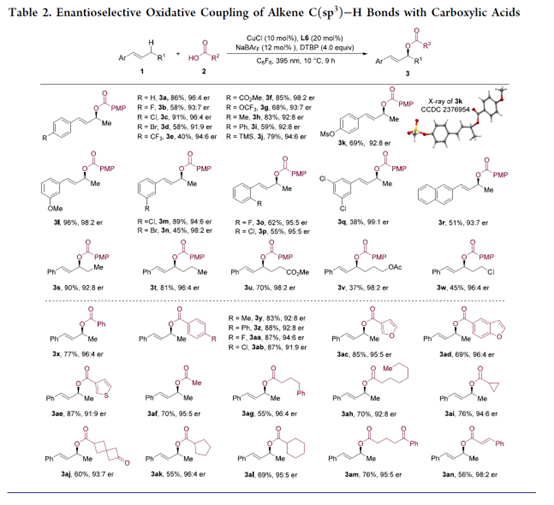
随后,作者对炔烃C(sp3)−H键与羧酸参与对映选择性氧化偶联反应的底物范围进行了扩展(Table 3)。通过对反应条件的优化后发现,一系列不同取代的(杂)芳基炔烃,均具有合理的收率与较高的对映选择性(5a–5r,91:9-97:3 er)。含有烷基链及烷氧羰基取代的底物,对反应对映选择性的影响可以忽略不计(5s–5v)。同样,脂肪族羧酸与不饱和羧酸,也是合适的底物,具有高度的对映选择性(如5w与5x)。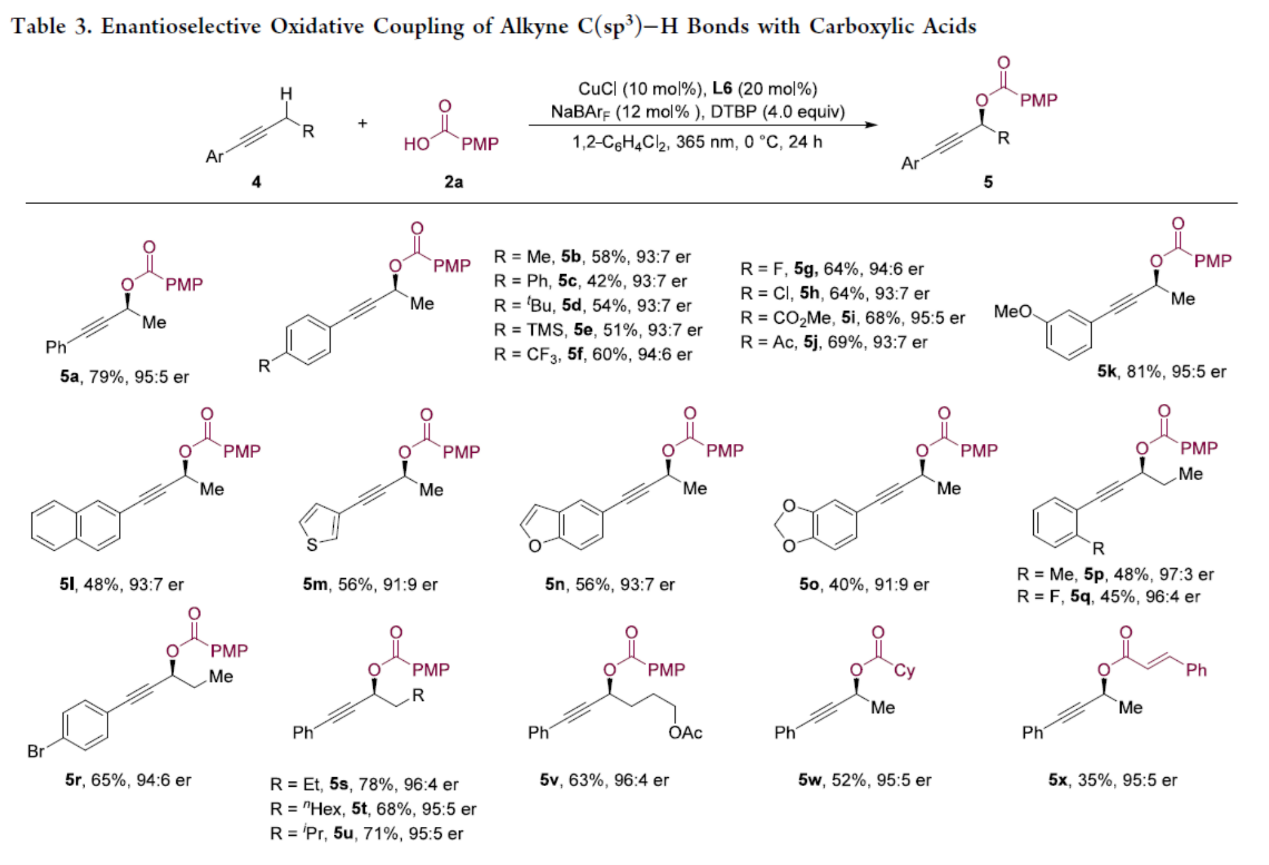
此外,该策略还可用于一些生物活性分子的后期衍生化,如布洛芬、萘普生、丙磺舒、吉非贝齐、熊果酸、玉米赤霉烯酮、Tigogenin与二氢胆固醇衍生物,具有高度的对映选择性,如3ao–3at、5y与5z(Figure 2)。这些例子强调了C(sp3)−H键与羧酸的对映选择性氧化偶联的高效性以及实用性。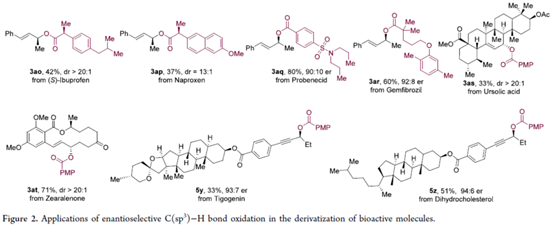
为了进一步了解反应的过程,作者对反应的机理进行了研究 (Figure 3)。自由基捕获实验结果表明,反应涉及自由基的机理(Figure 3A)。KIE实验结果表明,从C(sp3)−H键中攫取氢原子可能是一个限速步骤(Figure 3B)。铜-中间体的催化活性实验结果表明,反应涉及中间体Int-I与Int-II的形成过程(Figure 3C)。Int-I与Int-II的EPR测量以及原位制备的催化剂实验结果表明,反应过程涉及CuII中间体的形成(Figure 3D)。反应中Cu(III)-中间体的EPR测量结果表明,在395 nm光下存在一个禁阻跃迁(Figure 3E)。同时,反应可能存在CuIII离子。叔丁氧基自由基的EPR测量结果表明,叔丁氧基自由基主要是由DTBP的光分解产生(Figure 3F)。灯光开/关实验结果表明,表明该反应不涉及自由基链机理(Figure 3G)。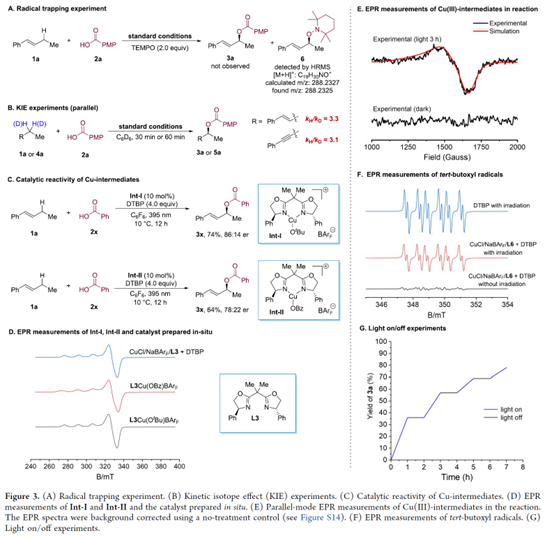
为了进一步深入了解反应的机理,作者还进行了相关的DFT计算研究 (Figure 4)。基于能量的计算,作者提出了一种合理的反应历程(Figure 4A)。首先,从CuI催化剂开始,与叔丁氧基自由基结合形成CuII-OtBu中间体Int-I,随后用羧酸取代配体形成CuII-OBz中间体Int-II,这两种反应都是放热反应。1a向CuII中心的自由基加成生成CuIII中间体Int-III,涉及开壳单重态(OSS)过渡态TS-III-OSS。然后,中间体Int-III通过TS-S-allylic-RA进行外层还原消除过程,形成Int-IV。最后,将另一个tBuO自由基加成到Int-IV中有助于其解离为目标产物3a,同时催化剂被再循环到Int-I中。其次,使用L6对上述提出机理的过渡态结构进行了研究(Figure 4B)。研究结果表明,过渡态结构中更强的空间排斥可能会提高对映选择性。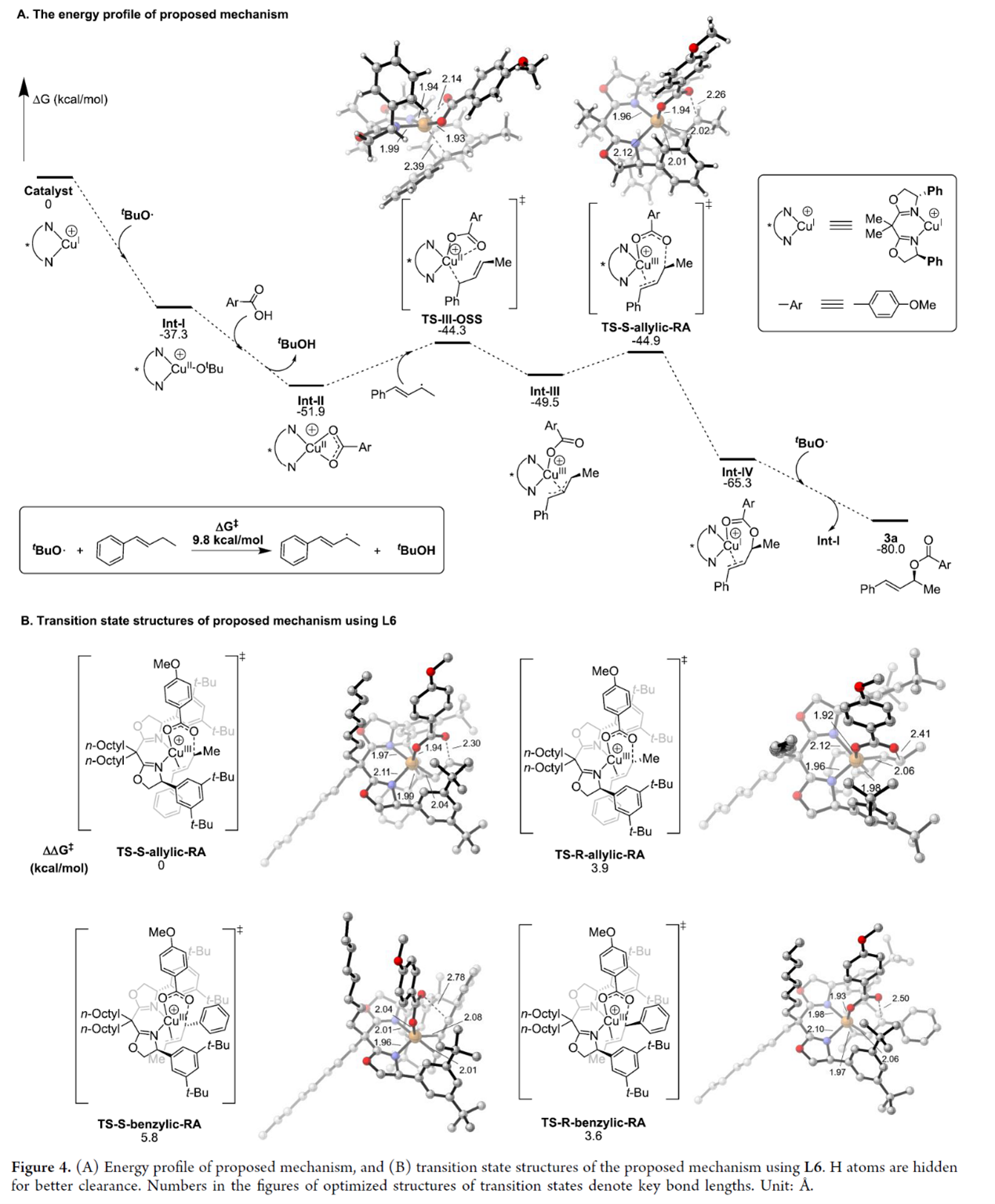
基于上述的讨论以及相关文献的查阅[1],作者提出了一种合理的反应机理(Figure 5)。首先,在辐照下,由DTBP分解生成叔丁氧基自由基,其可与CuI配合物反应形成CuII-OtBu中间体Int-I,并从底物中攫取一个氢原子形成烯丙基自由基。Int-I与羧酸发生配体交换反应,生成CuII-OBz中间体Int-II,后者与烯丙基自由基结合生成中间体Int-III。催化剂上的酰氧基与烯丙基通过分子内偶联,生成中间体Int-IV。最后,中间体Int-IV与叔丁氧基自由基反应生成烯丙酯并再生催化剂Int-I。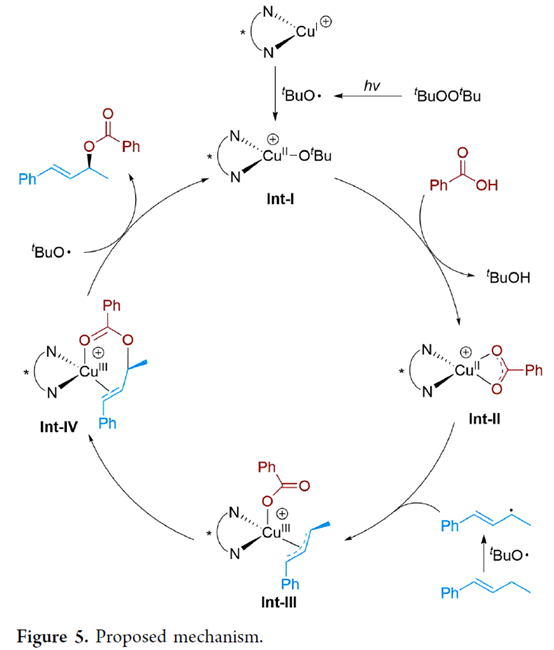
总结:
南开大学的周其林团队开发了一种阳离子铜催化方法,可以在蓝光下使烯丙基与炔丙基C(sp3)−H键与羧酸进行对映选择性氧化偶联反应。同时,该反应具有广泛的底物范围、良好的官能团耐受性、出色的对映选择性等优势。使用这种方法,可以直接从易于获得的C(sp3)−H底物合成各种手性酯。
参考文献:
[1] X. Chen, Z. Lian, S. Kramer, Angew. Chem., Int. Ed. 2023, 62, No. e202217638. doi:10.1002/anie.202217638. [2] J. M. Lee, E. J. Park, S. H. Cho, S. Chang, J. Am.Chem. Soc. 2008, 130, 7824. doi:10.1021/ja8031218. [3] M. S. Kharasch, G. Sosnovsky, J. Am. Chem. Soc.1958, 80, 756. doi:10.1021/ja01536a062.本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载.
关注Chem-Station抖音号:79473891841























No comments yet.