
作者:石油醚
导读:
近日,莫菲特癌症中心Justin Lopchuk课题组在Science Advances上发表论文,报道了通过可放大路线和后期官能团化策略实现复杂天然产物withanolides家族的发散性合成工作。该工作实现11个不同氧化程度withanolides天然产物的合成。步骤在11步至20步等,其中,灵活使用了一系列后期氧化策略包括烯丙基氧化等,尤其是仿生光氧化-烯丙基过氧化物重排反应,使得该路线较文献中已报道路线有明显缩短。该工作使得进一步的生物学研究能够脱离对天然产物母体或其简单衍生物的微量依赖。
“Divergent synthesis of complex withanolides enabledby a scalable route and late-stage functionalization.
Wen Che, Lukasz Wojitas, Chuan Shan, Justin M. Lopchuk*
Sci. Adv., 2024, 10, eadp9375. Doi: 10.1126/sciadv.adp9375”
正文:
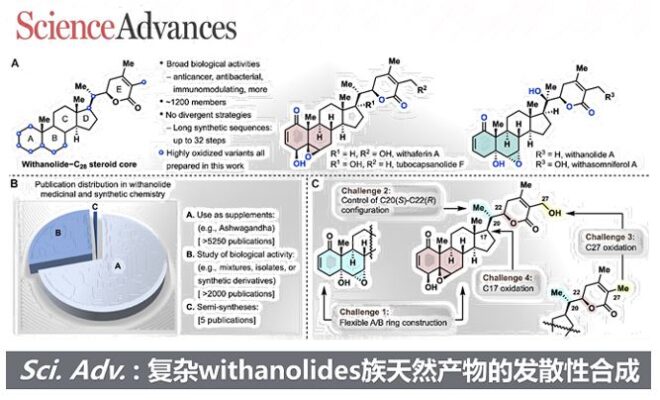
几个世纪以来,人类一直依赖自然界作为满足基本需求的可靠来源,包括提供应对各种疾病所需的药物。Withania somnifera的提取物——其梵文名称为“Ashwagandha”,也称为“印度人参”或“冬樱”——在印度传统医学体系阿育吠陀中已经使用了超过3000年。这种提取物的许多治疗特性可能与其中的withanolides有关。自1962年Lavie首次分离出第一个withanolide,withaferin A以来,这一类天然产物引起了生物学家和化学家的极大兴趣。withanolides是一组基于ergostane骨架的天然C28类甾体,其中C22/C26或C23/C26被氧化形成δ-或γ-内酯。生产withanolides的植物的一个特征是它们具有在环状骨架和侧链的几乎每个位置引入氧的非凡能力 (图1A)。这些复杂的立体化学氧化模式导致了多种生物活性,包括抗炎、抗肿瘤、免疫调节、癌症化学预防、抗菌和抗真菌特性。此外,据报道一些withanolides有可能被安全地用于COVID-19的预防和治疗干预措施。
鉴于上述广泛的生物活性,已有大量文献描述了withanolides在健康补充剂中的用途(超过5250篇出版物)或它们的生物活性,包括天然产物本身及其衍生物(超过2000篇出版物)(图1B)。相反,该家族合成仅有五篇半合成方法被报道,且没有一种被证明是发散性的或可扩展的:jaborosalactones A、B和D;withaferin A和27-脱氧withaferin A;withanolide D;withanolide E;以及withanolide A。还有各种关于片段结构构建和衍生化的报道。由于合成地限制,几乎所有用于生物或临床研究的主要材料都是从天然来源获得的,这不仅涉及耗时的提取和分离过程,还限制了超越简单衍生化的有效药物化学结构的发现研究。创建一种可扩展的、发散性的和灵活的withanolides合成路径,不仅能解决供应问题,还能便捷地获得一系列非天然类似物以进行生物活性研究。
withanolides的合成挑战包括:(i) 构建高度氧化的可变A/B环并同时组装内酯侧链;(ii) 控制整个分子尤其是δ-内酯构建中邻近C20(S)-C22(R)构型的相对立体化学;(iii) 引入C27羟基,这在先前的withaferin A合成中导致了较长的合成路线[1];以及(iv) 在C17选择性和立体选择性地引入羟基,从而使合成tubocapsanolides(图1C)变为可能。从逆合成的角度来看,后期的氧化将大大缩短C27羟基的引入步骤(图1D)。我们推测withanolides中C27的氧化很可能在自然界中通过O2的氧化和阳光下的重排发生。基于这种生物启发的假设,作者设想了一个Schenck ene-烯丙基过氧化物重排反应串联。为了实现发散性合成,需要通过不同的氧化反应在C4-C7上以区域和立体化学控制从一个共同的可拓展的中间体(本身为天然产物)获得两种经典的A/B环系统(1-氧-2-烯-4β-羟基-5β,6β-环氧,红色阴影和1-氧-2-烯-5α-羟基-6α,7α-环氧,蓝色阴影)(图1D)。最后,通过一系列精心设计的氧化反应和Vinylogous Aldo-环化反应,从廉价原料pregnenolone出发,可拓展中间体14。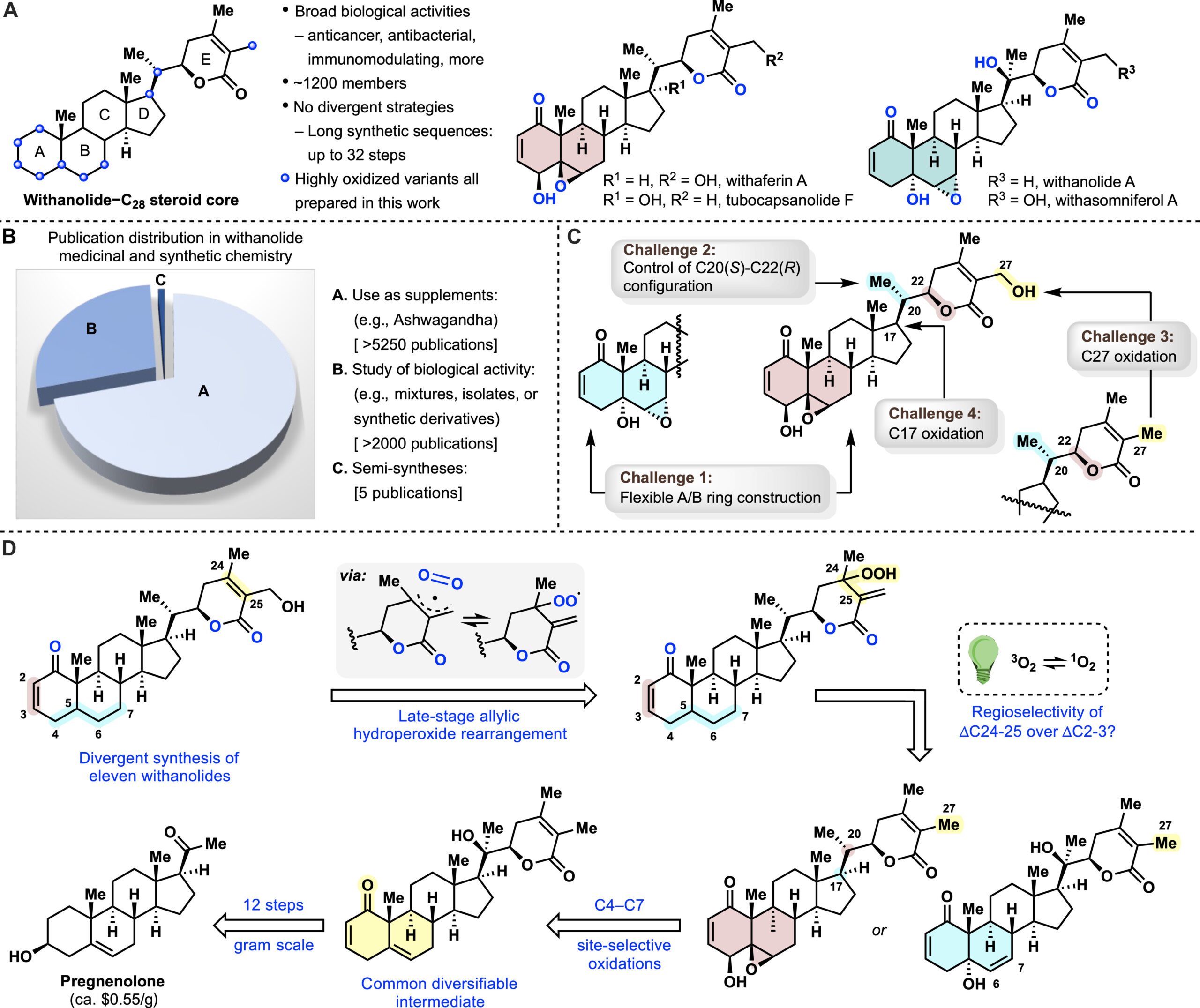 图1 withanolide族天然产物合成策略、挑战及其重要性。来源于Sci. Adv.
图1 withanolide族天然产物合成策略、挑战及其重要性。来源于Sci. Adv.
为了实现这一目标,需要开发出一种实用且可放大的路线,以获取天然产物14和withanolide D (16)(图2)。该路线以无保护基团自由的1,3-二噻烷对pregnenolone(1)的非对映选择性1,2-加成为起点,该反应可放大到10克规模。通过Oppenauer氧化二醇2,几乎定量地得到烯酮3。用氯甲基甲醚(MOMCl)保护叔醇3,得到烯酮4(62%产率)和全保护产物5(32%产率),后者经盐酸处理后,可以以80%的产率回收为3。经过一轮5回收的循环,从3得到烯酮4,总体产率为83%。随后作者筛选了各种氧化条件(DDQ、IBX和Saegusa氧化),最终,Mukaiyama脱氢以87%的产率生成6,并为C4-C5双键向C5-C6的动力学迁移奠定了基础。该烯烃异构化对反应时间敏感,较长的反应时间(例如1小时)会导致由于A芳构化产物的形成而使产率降低。Luche还原以>20:1的非对映选择性生成醇8。经过在不同中间体尝试引入侧链,确定需要在进一步引入电亲中心之前在中间体8上安装侧链。低温下用N-溴代丁二酰亚胺(NBS)处理,氧化脱除1,3-二硫烷,形成醛9而不影响A环中的烯丙醇。从2,3-二甲基丁-2-烯酸酯和六甲基二硅氮化锂生成的乙烯基醇酸盐与醛9反应,随后自发环化,得到内酯10,产率为78%。值得注意的是,这一过程中不需要保护A环的烯丙醇。内酯形成后,在2-硝基苯硒氰酸酯和三丁基膦条件下,10转化为硒化物11,产率为94%。在间氯过氧苯甲酸(mCPBA)条件下进行的Se-Mislow–Evans重排,随后在同一反应器中用甲醇钠(NaOMe)处理,得到12,产率为74%。Dess-Martin氧化和酸性条件下MOM基团的脱保护生成了共同的可多样化中间体(及天然产物)14,经过12步反应在多克规模上制备(单次产量为2.0克)。这个三步序列仅在最后一步后需要一次纯化,从硒化物11起总体产率为79%,得到烯酮14。接下来需要化学和立体选择性地引入C4 β-羟基,而不在B环或内酯上的四个其他烯丙基位置发生反应。经过仔细优化,发现二氧六环中的二氧化硒能提供单一立体选择性和最高的区域选择性。醇15在克规模下分离,产率为71%(基于回收的起始材料,产率为92%)。使用二氧六环(而非二氯甲烷)并在完全转化前停止反应,避免了不需要的C6氧化异构体。接下来,作者尝试使用Ikekawa的方法,在mCPBA条件下进行环氧化。然而,作者发现在大规模反应中,该条件下C5-C6环氧化物的比例为2:1(β:α)。相反,钛介导的环氧化反应以75%的产率单一非对映体获得withanolide D(16),且未观察到C2─C3烯烃的反应。这个14步的可放大路线可以单次制备919 mg的withanolide D,为即将进行的发散性合成奠定基础。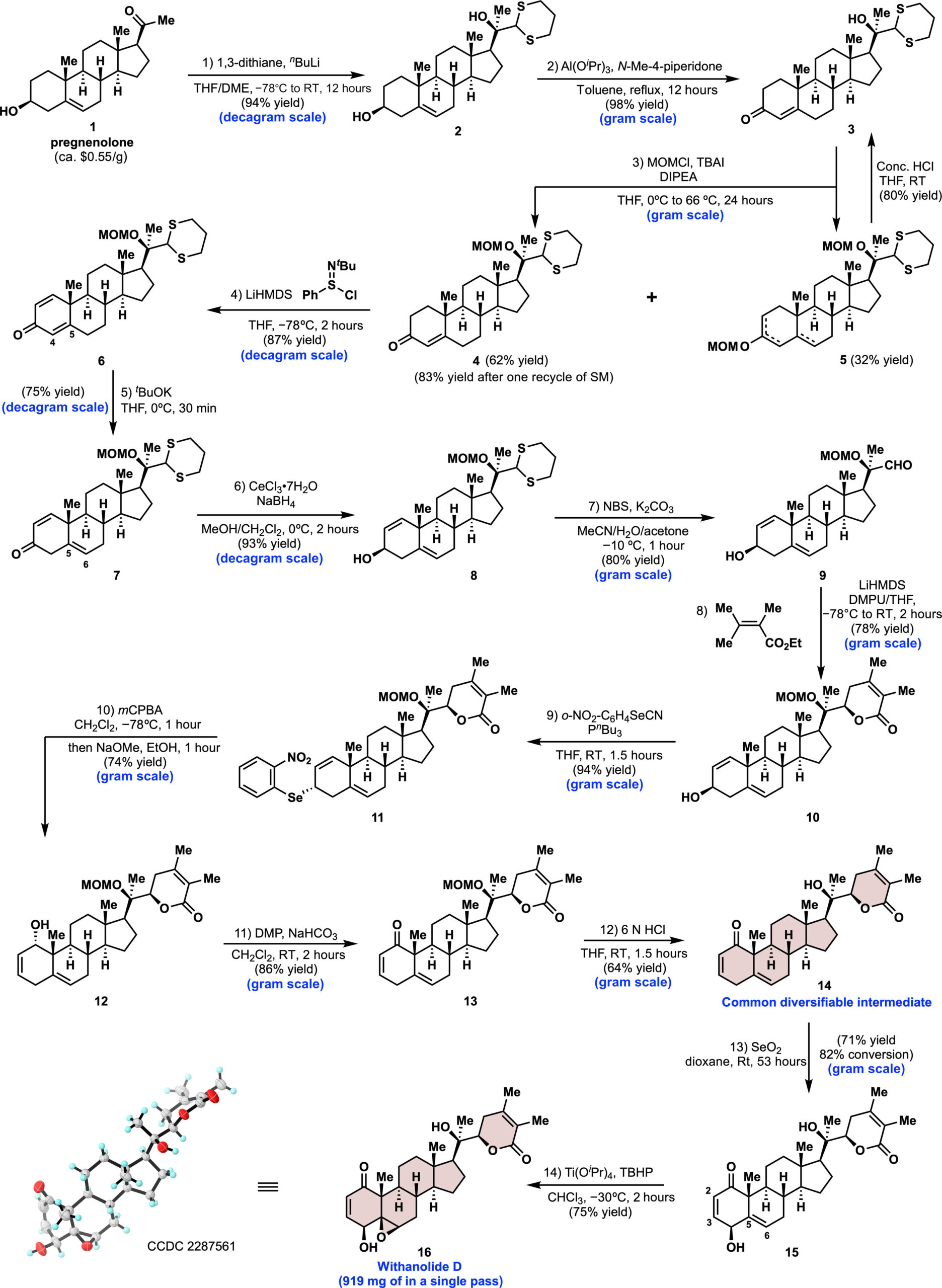
图2 withanolide D及其中间体的合成。来源于Sci. Adv.
在成功建立了通向关键中间体14和withanolide D (16)的实用路线后,我们继续合成了其他九种withanolides。通过CeCl3•7H2O进行区域选择性的环氧开环反应,将withanolide D (16)转化为17,产率为86%。使用碳负载的钯在常压下选择性地氢化C2─C3双键,得到2,3-二氢withanolide D (18) ,产率为93%(图3A)。C27的氧化状态是天然withanolides中常见的修饰。因此,一个关键挑战是在存在大量敏感功能团和多个竞争氧化位点的情况下,对C27进行后期氧化。因为C27既是内酯的β-位置又是烯丙位,最初尝试在NBS与AIBN或光照的自由基条件下进行。然而,尽管进行了许多优化,结果仍是得到复杂的混合物。受到Foote和同事工作的启发[2],我们转而尝试α,β-不饱和内酯的光氧化。首先,用三乙基硅基(TES)保护C4 β-羟基,得到19,产率为93%。在绿色LED光下使用Rose bengal为催化剂的Schenck ene条件处理19,作者发现起始原料显著转化。反应混合物分析揭示了三个主要产物:氢过氧化物21、氧添加产物22和所需的C27氧化产物26。反应需要3天才能完全转化,在此期间,氢过氧化物21逐渐转化为22和26。尽管对烯烃和烯醇醚的光敏化氧化反应已经有广泛研究[3],但涉及不饱和酮或酯的报道却很少(29),特别是在天然产物合成的背景下。我们推测,通过适当的烯丙醇转位方法,21和22都可以分离并转化为所需的产物26。吡啶二氯铬酸盐(PDC)介导的烯丙基转位反应使21和22分别以53%和71%的产率转化为26。精确控制PDC的用量(1.1倍当量)避免了醇氧化为相应的醛。从19到C27羟基26的总组合产率为61%(图3B)。该反应的机制可能包括两个阶段(图3C)。单线态氧与19内酯中的缺电子烯烃反应,形成过氧化物中间体20,随后夺取β-质子,生成氢过氧化物中间体21[3]。烯丙基氢过氧化物的重排涉及一个笼状烯丙基自由基-双氧对过渡态,随后进行[3C,2O] σ-迁移,生成重排的氢过氧化物产物25[4,5]。同样的22到26的整体转化通过PDC的铬酸酯实现(图3C)。随后作者开发了一种简化的一锅法,其中19依次经过光氧化条件、二甲基硫(DMS)和PDC,直接得到26,产率为52%。在成功氧化C27位置后,在常规四丁基氟化铵(TBAF)条件下尝试去除TES基团,结果导致原料分解。最终确定TBAF溶液的碱性是有害的。最终作者发现,用醋酸缓冲的TBAF处理26,可以得到27-羟基withanolide D (29),产率为81%。
图3 withanolides的发散性全合成及C27后期氧化. 来源于Sci. Adv.
在确定了withanolide D的A/B环的氧化路线和C27后期羟基化之后,我们将注意力转向含有另一种A/B环结构的其他代表性withanolides(以withanolide A为例),进一步突显了易得中间体14的多功能性(图4)。关键转化是C5 α-羟基的引入。受Gademann开创性工作的启发[6],作者将共同中间体14置于吡啶作为溶剂的光氧化条件下,使用白色LED灯,得到withacoagin(30),产率为40%。尝试进一步优化氧化条件,包括更换溶剂、光催化剂、光源以及改变还原剂,都没有显著提高产率。副产物被鉴定为C2和C5之间形成了过氧桥;然而,对没有C2─C3烯烃的起始材料进行光氧化结果产生复杂的混合物,没有找到所需产物。然后用异丙醇钛和叔丁基氢过氧化物处理withacoagin,立体选择性地得到的withanolide A (31),产率为70%。然后将withanolide A (31)在不进行保护的情况下,置于合成27-羟基withanolide D (29)时开发的Schenck ene-重排条件下,得到84%产率的氢过氧化物32、醇33和所需的天然产物withasomniferol A (34)。与19到26的转化类似,氢过氧化物32首先形成,并逐渐转化为33和34。分别用DMS处理过氧化物32和醇33,随后用PDC处理,得到withasomniferol A (34),总产率为53%。值得注意的是,withasomniferol A的制备从14开始,没有使用保护基,这突显了仿生C27氧化方法的温和和有效性。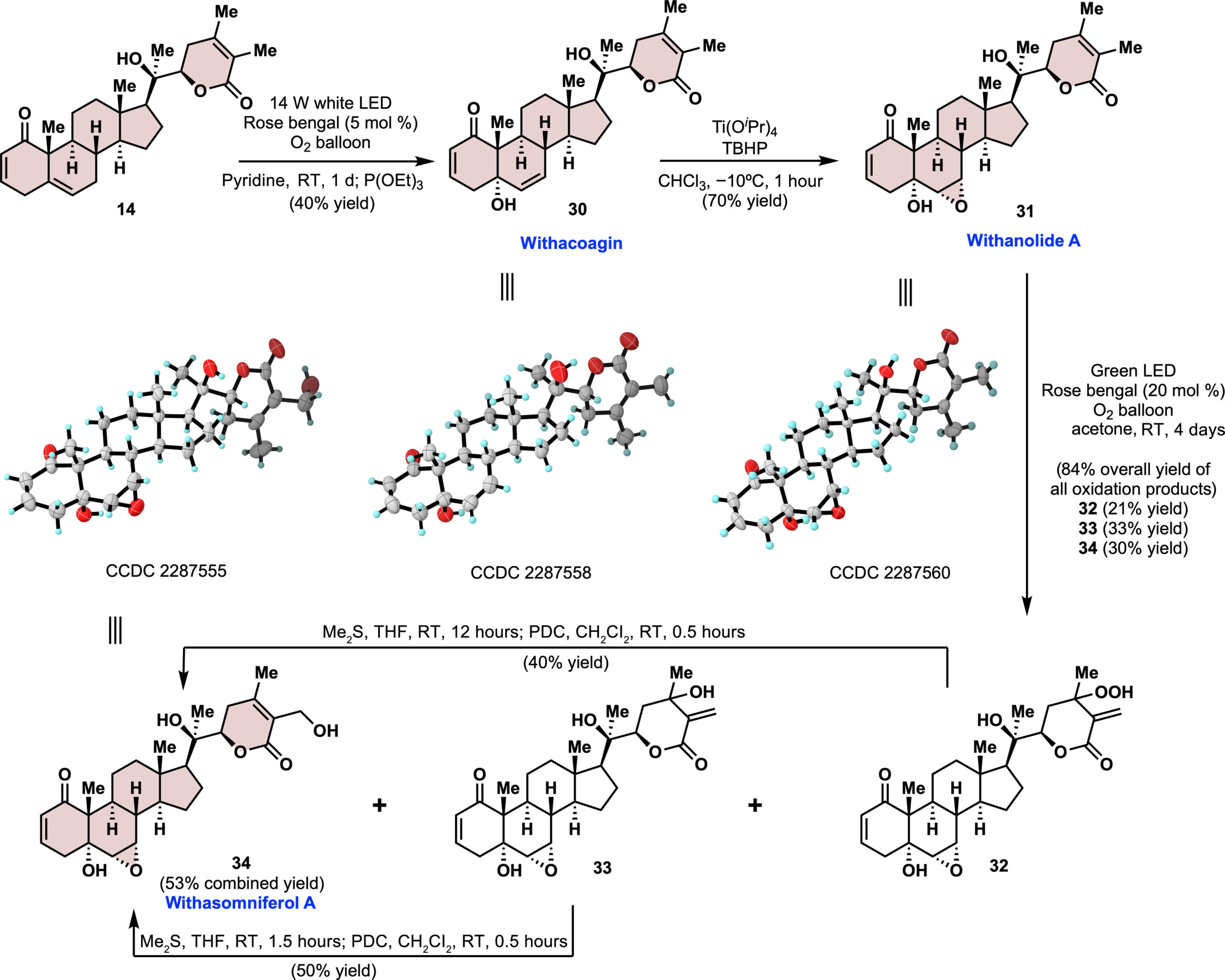
图4 withacoagin、withanolide A和 withasomniferol A的合成. 来源于Sci. Adv.
withaferin A由于其具有潜在的治疗前景,吸引了发现化学家们的广泛兴趣。withaferin A最具前景的应用之一是其抗癌和化学预防潜力,已有来自多个团队的多项研究证实[7]。尽管如此,Ikekawa等人的32步合成方法仍然是已知唯一的报道[1]。在获得正确的A/B环结构和C27氧化之后,剩下的任务是去除C20羟基,同时保持剩余甲基正确的立体化学。通常,在具有显著空间位阻的位置上的进行脱氧反应是非常具有挑战性的;在这种情况下,在复杂的分子环境中进行这种操作就更增加了难度。为此,作者研究了多种脱氧方法。基于酸活化后还原淬灭的方案要么没有转化(在室温下)要么在高温下分解。自由基相关的脱氧策略由于如上所述,C20羟基官能团化由于位阻较大而显得困难。具体来说,将其转化为常规前体如黄原酸盐、硫羰基氨酸盐、m-CF3苯甲酸盐或N-邻苯二甲酰亚氨基草酸酯衍生物的尝试均未成功——所有结果要么没有转化要么是起始原料分解。面对这些失败,我们选择了两步法:消除和还原。用吡啶和氯化亚砜(SOCl2)的初步尝试导致起始原料分解,而Burgess试剂条件下原料未发生转化。而使用Martin sulfurane, 原料在1小时内由TES保护的withanolide D 19生成了exo-双键产物35,产率为86%(图5A)。在获得35后,作者研究了不同的氢化催化剂。钯碳催化剂、均相Crabtree催化剂和Wilkinson催化剂导致了C2─C3双键的还原,而C20-C21烯烃保持不变。各种金属氢化物原子转移反应要么回收了起始材料,要么导致分解。令作者高兴的是,二氧化铂作为催化剂条件下产生了目标产物36,产率为75%,非对映体比率为2:1。C2─C3双键首先还原,大约在6小时内,而C20-C21烯烃反应较慢。用溶剂(甲醇和乙酸乙酯)或添加剂改进非对映体比率的尝试未能成功。重新生成C2─C3不饱和键比预期更为艰难:先前使用的Mukaiyama脱氢反应失败,IBX处理要么没有转化要么在不同温度下导致B环环氧裂解。意外地,用苯硒酸酐处理36得到的酮37,产率为88%。使用TBAF进行的一锅法脱保护提供了27-脱氧withaferin A(38),从36到38的一锅法产率为73%。将TES保护的27-脱氧withaferin A 37暴露于Schenck ene-重排条件下,通过前面描述的相同路径得到TES保护的withaferin A 41,总体产率为63%。用缓冲TBAF溶液脱保护完成了withaferin A(41)的合成。Tubocapsanolide F首次在2007年被分离出来,代表了withanolides的一个亚族,其中C16和C17均被氧化[8]。它对多种肝癌、乳腺癌和肺癌细胞系表现出细胞毒活性。withanolides及相关骨架的C16和C17 C─H键由于缺乏有效手段而往往难以官能团化,尽管Breslow的远程功能化方法代表了这种问题的一种替代解决方案[9]。在此,我们利用中间体35的分支路线来制备tubocapsanolide F(图5B)。将35暴露于60 ℃的二氧六环中的二氧化硒,导致C17的位点选择性和立体特异性氧化,得到烯丙醇43,产率为76%。在常压下使用二氧化铂进行氢化生成了产物44,产率为77%(非对映体比率为2:1)。C2─C3双键以60%的产率重新生成,随后脱保护得到tubocapsanolide F。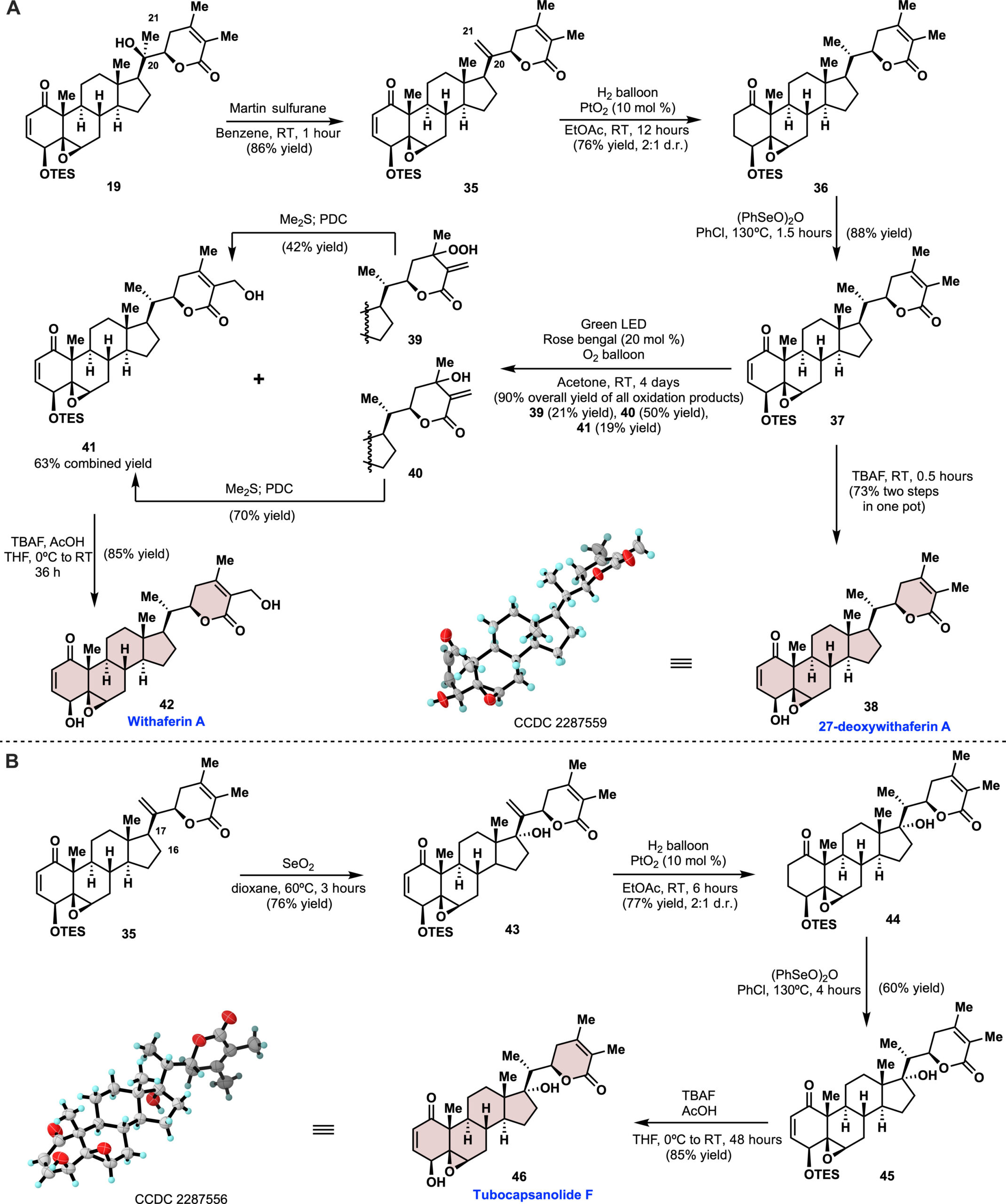
图5. withaferin A及 C17氧化路线合成tubocapsanolide F. 图来源于Sci. Adv.
总结:
莫菲特癌症中心的Justin Lopchuk 课题组报道的克级规模的路线使得同时合成一个共同的中间体和withanolide D成为可能,从而在12到20步内合成了11种withanolides,并且有八种结构通过X-ray射线晶体学确认。通过选择性烯丙基氧化和光化学Schenck ene反应,制备了两种经典的A/B环结构,代表性化合物为withaferin A和withanolide A。生物启发的光化学氧化-烯丙基过氧化物重排串联实现了后期的C27 C─H氧化,相较于之前的报道,显著提高了合成步骤的经济性。C17的位点和立体选择性烯丙基氧化实现了D环的后期功能化,而无需使用预先安装的模板导向基团。该路线的简洁性、可扩展性、模块化和以多样性为导向的特性预计将提供多种withanolides,并允许有针对性地制备更适合的非天然类似物,为开发治疗药物提供了广阔的前景。
(感谢Justin Lopchuk对Chem-Station的支持,感谢文章作者车稳提供稿件)
参考文献:
- [1] M. hirayama, K. Gamoh, N. ikekawa, Stereoselective synthesis of withafein A and27-deoxywithaferin A1. Tetrahedron Lett. 1982, 23, 4725–4728.
- [2] B.-M. Kwon, R. C. Kanner, C. S. Foote, Reaction of singlet oxygen with 2-cyclopenten-1-ones. Tetrahedron Lett. 1989, 30, 903–906.
- [3] M. Prein, W. Adam, the Schenck ene reaction: diastereoselective oxyfunctionalizationwith singlet oxygen in synthetic applications. Angew. Chem. Int. Ed. Engl. 1996, 35, 477–494.
- [4] A. G. davies, the Schenck rearrangement of allylic hydroperoxides. J. Chem. Res. 2009, 533–544.
- [5] A. L. J. Beckwith, A. G. davies, I. G. E. Davison, A. Maccoll, M. H. Mruzek, The mechanisms of the rearrangements of allylic hydroperoxides: 5α-hydroperoxy-3β-hydroxycholest-6-ene and 7α-hydroperoxy-3β-hydroxycholest-5-ene. J. Chem. Soc. Perkin Trans.2.1989, 2, 815–824.
- [6] C. K. Jana, J. hoecker, T. M. Woods, H. J. Jessen, M. neuburger, K. Gademann, Synthesis ofwithanolide A, biological evaluation of its neuritogenic properties, and studies onsecretase inhibition. Angew. Chem. Int. Ed. EngL. 2011 50, 8407–8411.
- [7] T. Sultana, M. K. Okla, M. Ahmed, N. Akhtar, A. Al-hashimi, H. Abdelgawad, Ihsan-ul-Hag,Withaferin A: From ancient remedy to potential drug candidate. Molecules, 2021, 26, 7696.
- [8] P.-W. hsieh, Z.-Y. huang, J.- H. Chen, F.-R. Chang, C.-C. Wu, Y.- L. Yang, M. Y. Chiang, M.- H. Yen, S.- L. Chen, h.-F. Yen, T. lübken, W.-C. Hung, Y.-C. Wu, Cytotoxic withanolidesfrom Tubocapsicum anomalum. J. Nat. Prod. 2017, 70, 747–753.
- [9] P. B. Reese, Remote functionalization reactions in steroids. Steroids 2001, 66, 481–497





















No comments yet.