
本文作者:有机小白
导读
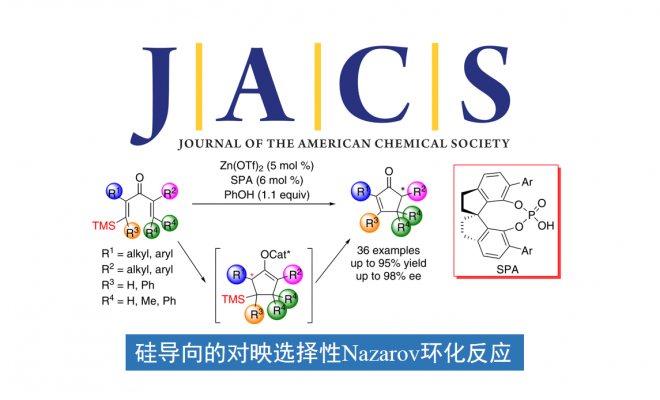
Nazarov环化,是一种经典的4π电环化反应,被广泛用于构筑环戊烯酮。在过去的几十年中,其不对称催化反应得到了广泛地研究,但控制双键位置的策略受限于产物取代基的影响,大大限制了Nazarov环化在有机合成上的应用。在此,南开大学化学学院与元素有机国家重点实验室朱守非教授团队报道了Lewis酸与手性Brønsted酸协同催化的高对映选择性硅导向的Nazarov环化反应。机理研究表明,Lewis酸和Brønsted酸的结合共同活化了二烯酮底物,反应的对映选择性来源于手性Brønsted酸促进的烯醇中间体的质子转移反应。
Enantioselective Silicon-Directed Nazarov Cyclization
Jin Cao, Meng-Yang Hu, Si-Yuan Liu, Xin-Yu Zhang, Shou-Fei Zhu,* and Qi-Lin Zhou
J.Am. Chem. Soc.2021, ASAP. DOI:10.1021/jacs.1c01194.
正文
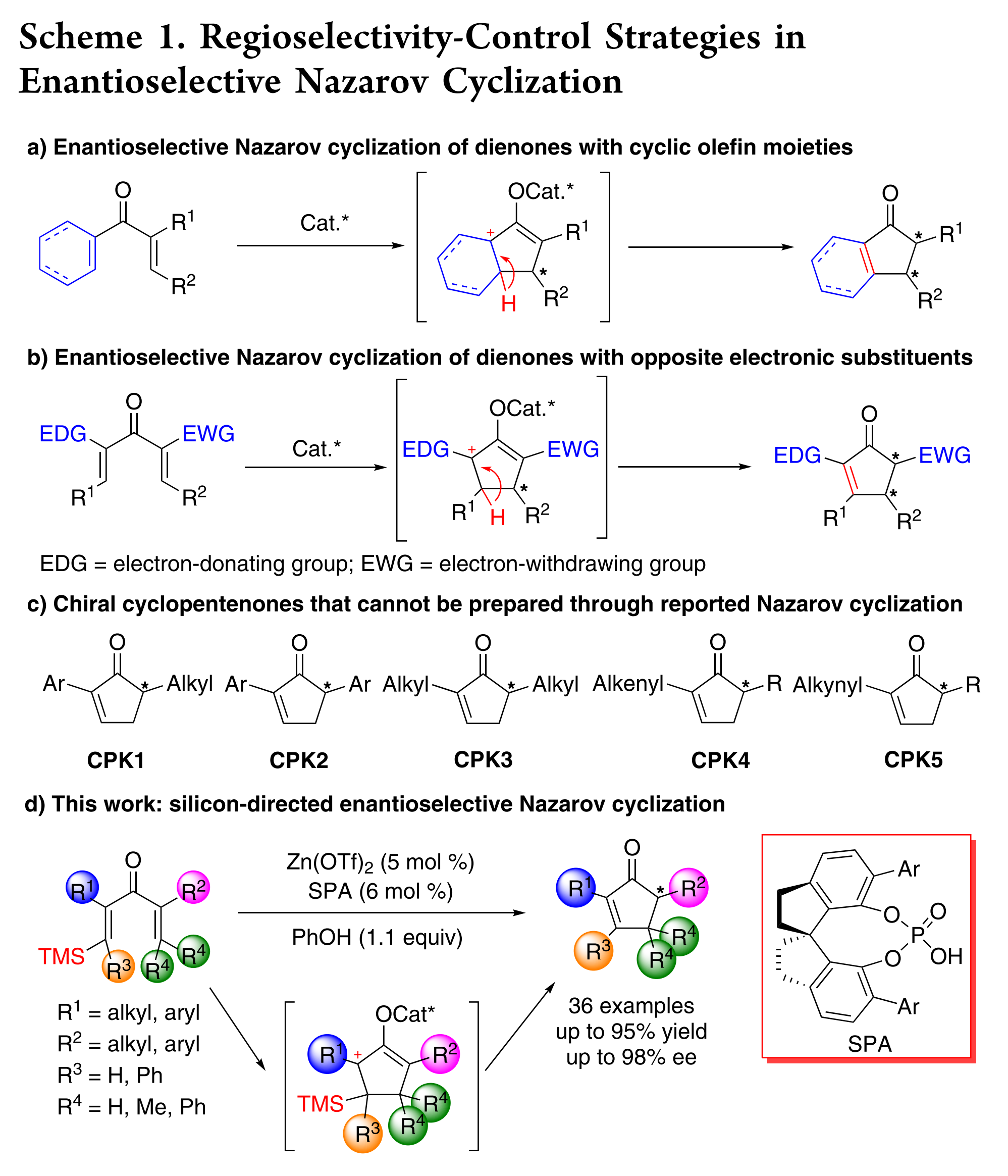
手性环戊烯酮是不对称合成中的关键中间体,其作为核心骨架广泛存在于许多天然产物和药物分子中。Nazarov环化,是一种经典的4π 电环化反应,能够一步合成多官能团的环戊烯酮。近几十年来,催化不对称Nazarov环化反应的研究已经取得了重要进展。然而,环化过程经过碳正离子中间体进行,该中间体易发生重排,因此,底物分子中含有多个可进行消除的β-氢时,会产生双键位置不同的环化产物。目前,化学家们已发展许多策略来选择性控制双键的位置。例如:1)将环烯烃结构引入二烯酮底物中,以增加某一种碳正离子的稳定性,从而控制双键在双环产物的位置(Scheme 1a);2)二烯酮的一个双键引入给电子基团,另一个双键引入吸电子基团,导致在给电子基团一侧产生碳正离子,从而通过电性来控制产物中双键的位置(Scheme 1b)。尽管上述策略取得了巨大的成功,但对底物取代基的要求,严重限制了所得产物的多样性。例如,这两种策略均不能对映选择性合成α,α’-二取代环戊烯酮(Scheme 1c),包括α-烷基-α’-芳基环戊烯酮(CPK1),α,α’-二烷基或α,α’-二芳基环戊烯酮(CPK2, CPK3),或α-烯基或α-炔基环戊烯酮(CPK4, CPK5)。因此,精确控制Nazarov环化反应的对映选择性,合成具有高度通用性的多官能化环戊烯酮是一项迫切需要且十分具有挑战性的研究课题。
1982年,Denmark和Jones报道了一种硅导向的Nazarov环化方法,截至目前,化学家对该方法进行了大量的研究。在该策略中,硅基团通过β-硅效应稳定烯酮底物中产生的碳正离子中间体,随后硅基消除,仅在目标位置形成双键。这种无痕的硅导向策略最大限度地减少了底物取代基的限制,从而为合成多种环戊烯酮提供了一种简洁、有效的方法。进一步研究表明,Sn和F也可以作为Nazarov环化反应的导向基团。然而,硅导向的Nazarov环化反应通常需要使用化学计量或亚化学计量的强Lewis酸或Brønsted酸作为催化剂,导致该反应的对映选择性反应还没有得到发展。在此,南开大学化学学院与元素有机国家重点实验室朱守非教授团队通过使用由非手性Lewis酸和手性Brønsted酸组成的协同催化体系,实现了硅导向高对映选择性的Nazarov环化反应(Scheme 1d)。该催化体系显著提高了反应速率,且能够有效地控制对映选择性,合成了具有前所未有的结构多样性的手性环戊烯酮,这是其他催化对映选择性反应(包括以前报道的对映选择性Nazarov环化反应)所没有实现的。
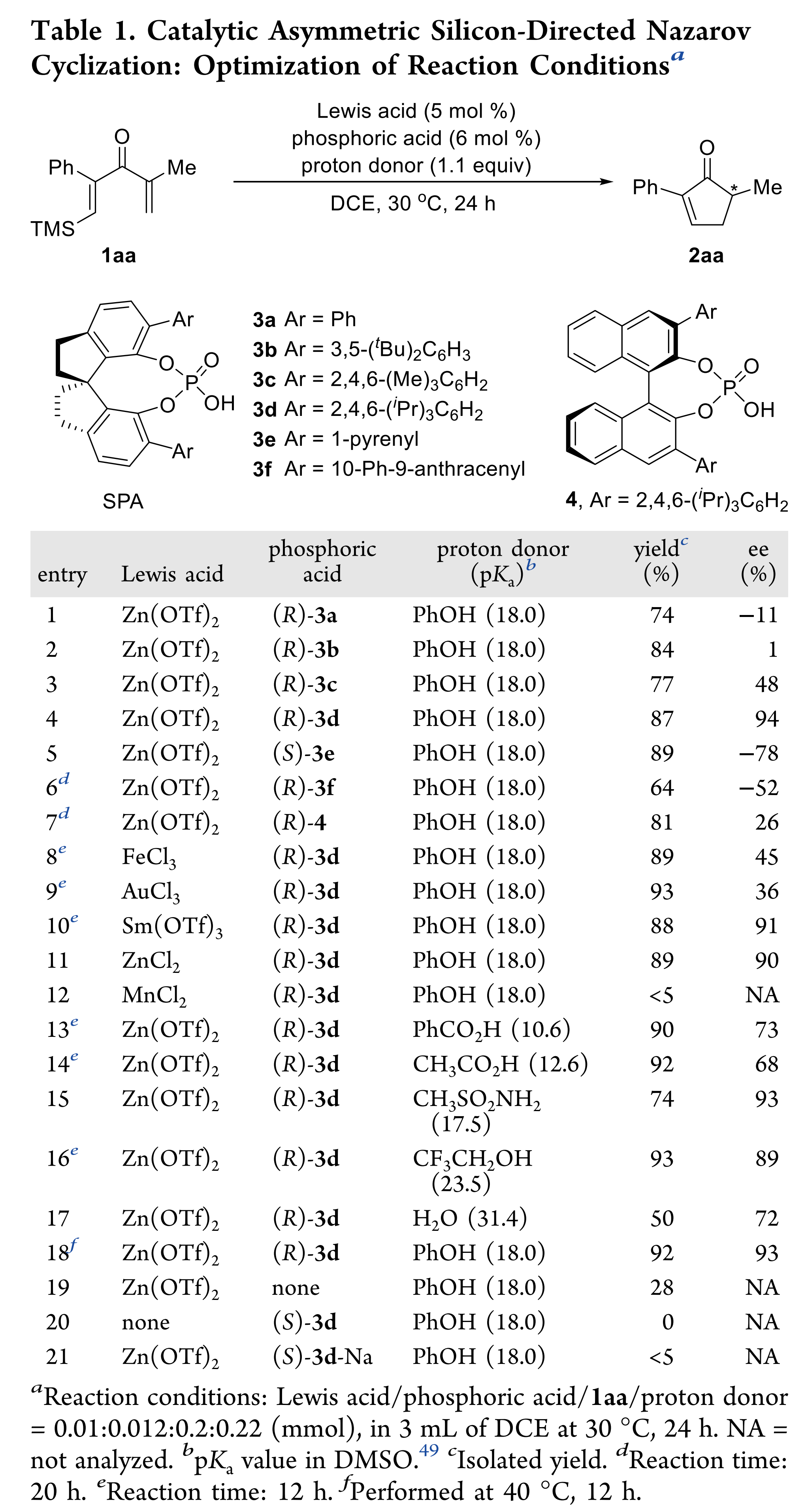
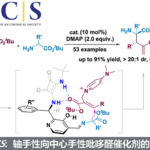
首先,作者以含有TMS取代基的二烯酮1aa作为模板底物,进行了反应条件的优化(Table 1)。最终,最优反应条件确定为:Lewis酸Zn(OTf)2与Brønsted酸(R)-3d作为协同催化剂,PhOH作为质子给体,在DCE中40 °C反应,以92%的收率和93% ee得到了目标产物2aa(entry 18)。控制实验表明,螺环磷酸对反应具有明显的促进作用(entries 19 and 20)。当使用螺环磷酸的钠盐时,仅有痕量2aa产物(entry 21),这个结果说明螺环磷酸形成锌盐的过程被干扰,由于位阻太大或者Lewis酸性太弱从而无法活化1aa。
在最优反应条件下,作者进行了底物范围的考察(Table 2)。R1为各种取代的苯基(R2固定为Me)时均得到了良好的收率和高对映选择性。其中,吸电子基的底物反应性较低,因此需要更强的Lewis酸、更高的温度以及更多的PhOH以得到满意的收率。R1为稠环或杂环时也能给出不错的结果。R1固定为苯基,当R2为直链或支链烷基、官能化烷基或苄基时,反应顺利进行,得到了满意的收率与对映选择性。其中,增加烷基链长度会降低产物的ee值。当R1、R2同为芳基或烷基时,也能与反应条件很好地兼容。甚至R1为烯基、炔基时,结果同样非常好。R3、R4为非氢取代基时,能够得到高产率和高ee值,但dr值不高,说明在该反应条件下β-位的立体化学不能很好地控制。增加R2、R4的差异对产率、ee值和dr值影响不大。

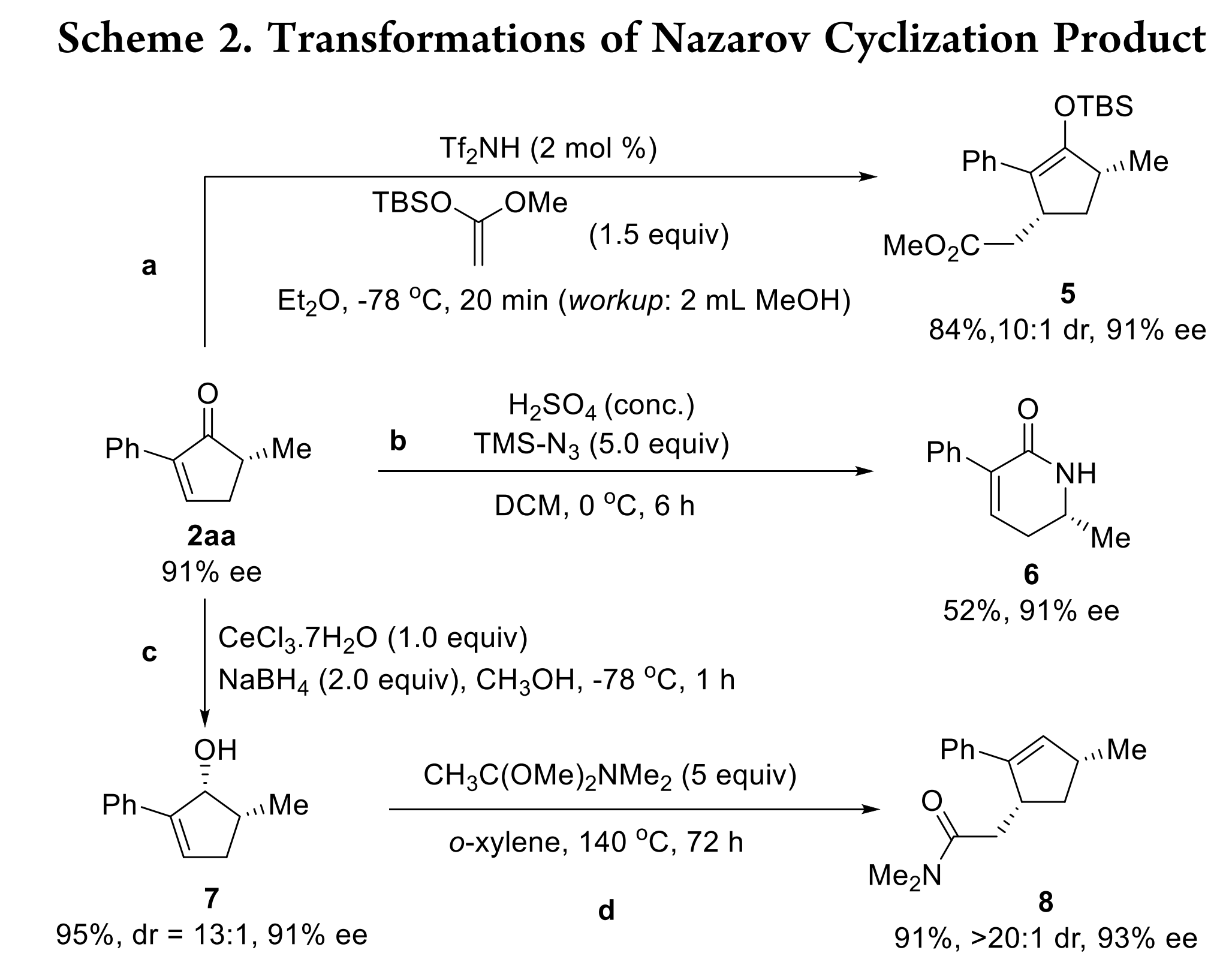
手性环戊烯酮骨架广泛存在于多种生物活性分子中,也是一种多功能的合成子。为了证明该反应的应用价值,作者对环戊烯酮2aa进行了转化实验(Scheme 2)。首先,将它作为Mukaiyama-Michael加成反应中的Michael受体,生成具有两个手性中心的稳定烯醇硅醚5,dr值很高,ee值也没有损失。通过Schmidt反应还将2aa转化为δ-内酰胺6,ee值也没有降低。此外,2aa可以以高dr值被还原为烯丙醇7,然后继续发生Eschenmoser-Claisen重排得到一个含有两个手性中心的产物(8)。
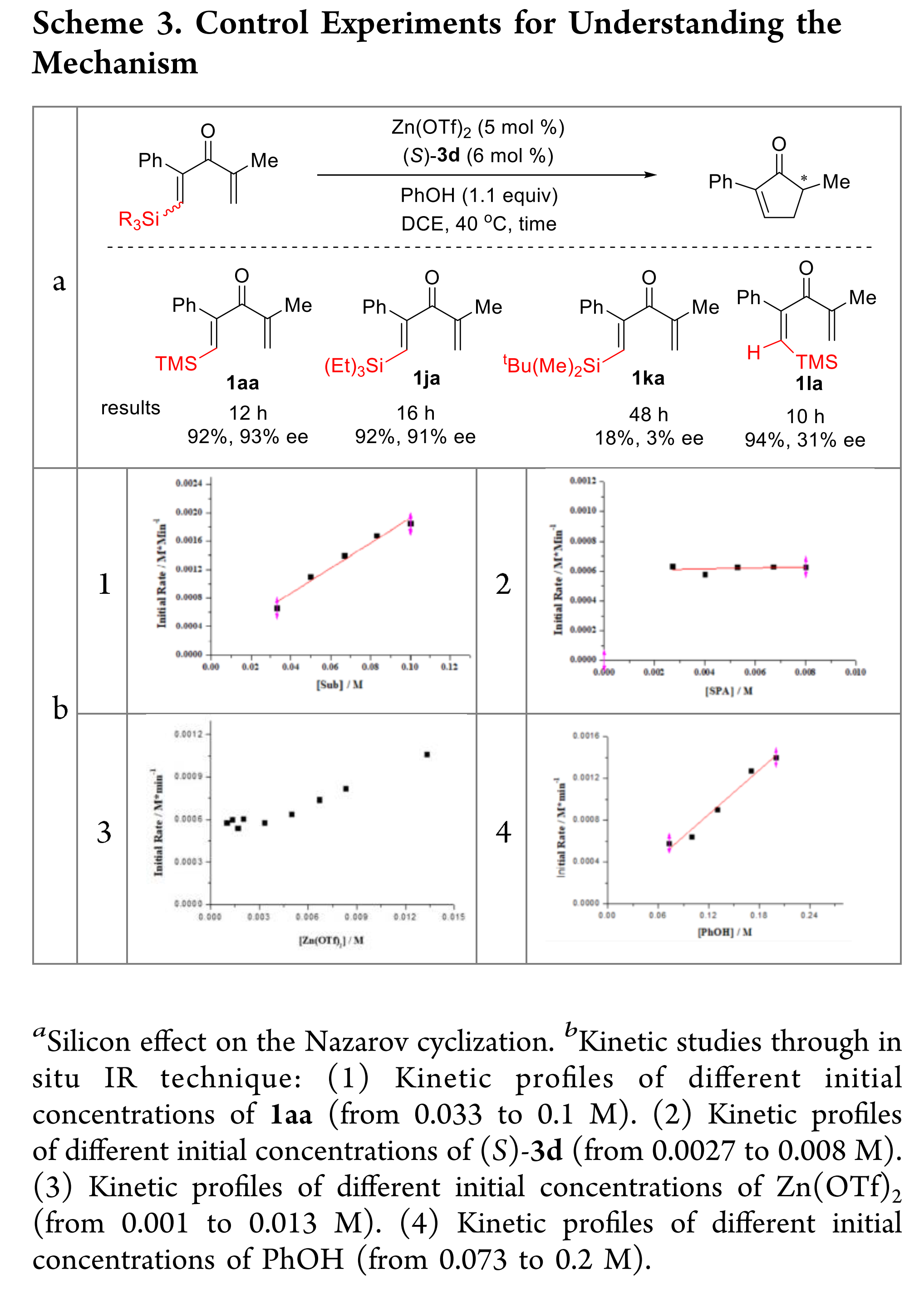
为了阐明反应机理,作者研究了底物上不同硅基团的影响(Scheme 3a)。增加硅基取代基的体积,明显降低了产率和对映选择性。硅基取代双键的构型也强烈影响对映选择性(1aa vs 1la, Scheme 3a)。作者通过改变反应物的浓度来研究反应动力学,通过原位红外光谱观察反应的初始速率(Scheme 3b)。反应对底物和苯酚均为一级反应。相比之下,该反应相对于手性螺环磷酸(R)-3d为零级反应。在较低的Zn(OTf)2浓度下,反应对该组分为零级反应,但随着Zn(OTf)2浓度的增加,反应明显加快。作者推测反应机理可能与Zn(OTf)2/(R)-3d的比例有关。当Zn(OTf)2浓度小于(R)-3d浓度时,底物和苯酚参与速率决定步骤(r = k[1aa][PhOH]),而当Zn(OTf)2浓度大于(R)-3d浓度时,底物、苯酚和Zn(OTf)2参与速率决定步骤(r = k[1aa][Zn(OTf)2][PhOH])。
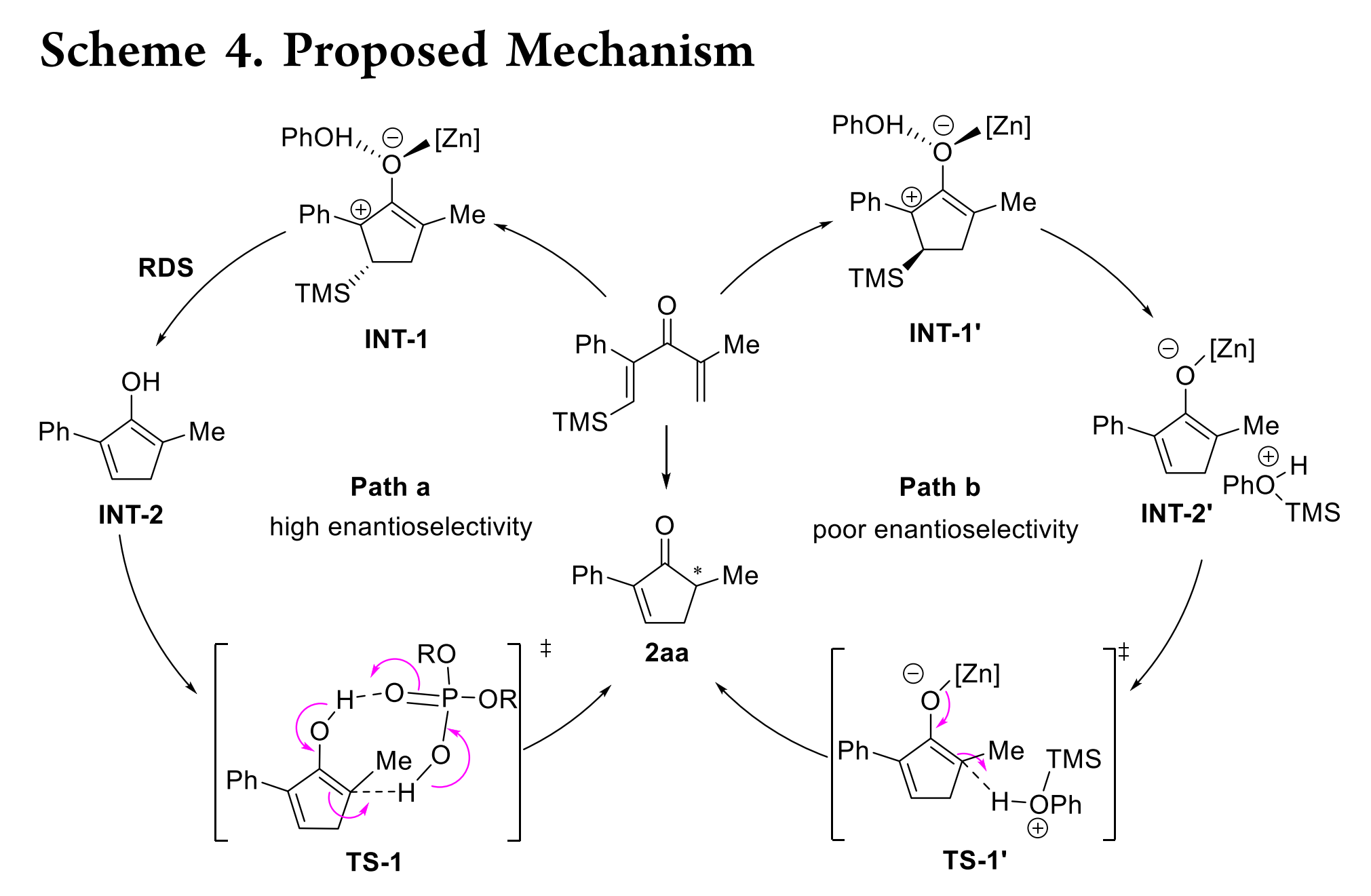
基于上述结果,作者提出了如Scheme 4所示的反应机理。首先,Zn(OTf)2和PhOH活化底物,使其发生4π 电环化得到碳正离子烯醇中间体INT-1。三甲基硅烷基对于配位的苯酚有两种可能的构型,分别如INT-1和INT-1’所示。在cis-构型(INT-1)中,可能发生配位苯酚对硅烷基的分子内进攻,产生自由烯醇中间体INT-2。最后,INT-2通过SPA质子化生成目标产物。在整个催化循环中,由于硅原子独特的电子性质,碳正离子仍然固定在与硅基相关的β-位,这是产生高度可控的区域选择性的根本原因。从INT-2生成目标产物的质子转移步骤是对映选择性决定步骤。在这一步中,手性SPA作为一个手性质子梭,促进了苯酚质子通过氢键向烯醇转移,从而控制立体化学(TS-1)。根据以上动力学研究,硅基团的消除可能是反应的决速步,且Zn(OTf)2和苯酚都参与了这一步骤。当SPA浓度超过Zn(OTf)2时,SPA可能通过络合作用促进Zn(OTf)2与INT-1的解离,从而在低浓度下观察到Zn(OTf)2为零级的反应动力学。尽管SPA在限速步骤中不直接促进反应,但它可能通过显著降低烯醇中间体质子转移的能垒来加速整个反应速率。然而,如INT-1’所示,当硅基与配位苯酚呈trans构型时,硅基的消除很可能由另一个苯酚协助,从而生成烯醇锌盐和苯酚的氧鎓。由于苯酚的氧鎓具有较高酸性的质子,可以在无SPA的辅助下(TS-1’)直接质子化烯醇锌盐,导致对映选择性低。不同的硅基及其初始构型可能导致INT-1与INT-1’的比率不同,因而这两种可能的途径可以用来解释硅基对该反应对映选择性的显著影响。由于反应机理的高度复杂性,需要对该反应进行更深入地研究。
总结
南开大学化学学院与元素有机国家重点实验室朱守非教授团队利用Lewis酸与Brønsted酸协同催化,实现了首例硅导向高对映选择性的Nazarov反应。在温和的反应条件下,高效合成了结构多样性、高区域选择性和高立体选择性的手性环戊烯酮。初步的机理研究表明,手性Brønsted酸通过氢键促进烯醇中间体的质子转移反应以实现不对称诱导。














No comments yet.