
本文作者:杉杉
导读
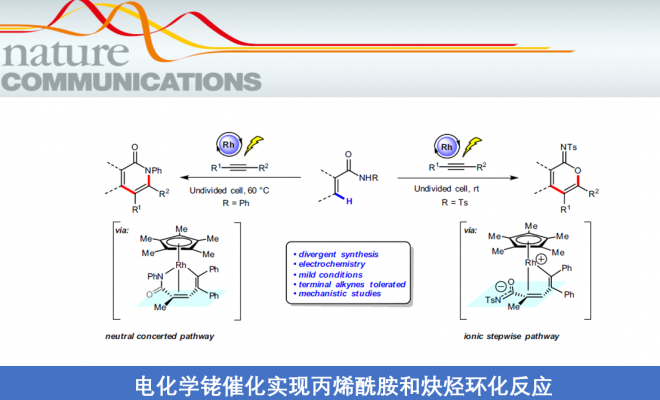
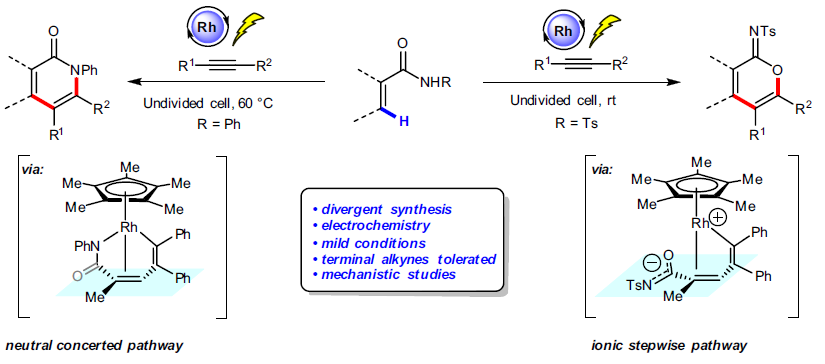
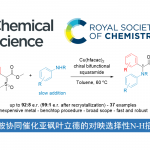
α-吡啶酮和α-吡喃酮是天然产物和生物活性分子中常见的骨架。近日,上海有机所梅天胜和浙江大学洪鑫课题组合作在Nat.Commun.上发表论文,报道了电化学Rh催化丙烯酰胺与炔烃的C-H环化反应,从而获得良好至优异收率的α-吡啶酮和α-吡啶酮化合物。同时,当使用不对称炔烃时,具有极好的区域选择性。其次,与传统过渡金属催化C-H环比相比,该方法可避免化学计量的金属氧化剂的使用。
Divergent rhodium-catalyzed electrochemical vinylic C-H annulation of acrylamides with alkynes
Yi-Kang Xing, Xin-Ran Chen, Qi-Liang Yang, Shuo-Qing Zhang, Hai-Ming Guo, Xin Hong &Tian-Sheng Mei
Nat.Commun. 2021, 12, 930. DOI:10.1038/s41467-021-21190-8
正文
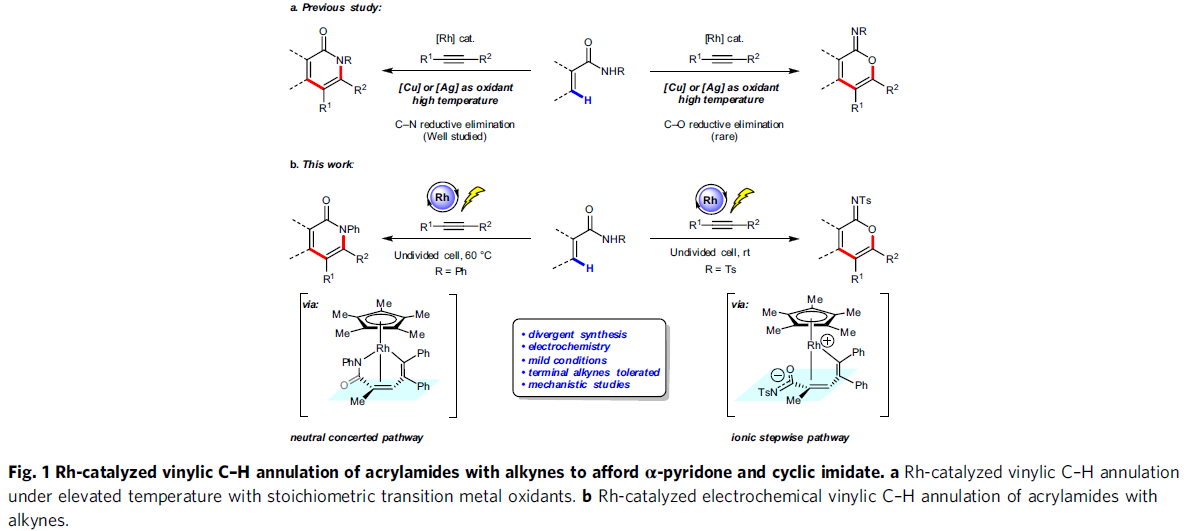
α-吡啶酮和α-吡喃酮是天然产物和生物活性分子中常见的骨架,而过渡金属催化丙烯酰胺或丙烯酸与炔烃的C-H环化反应是高效的合成工具。2009年,Miura等[1]报道了使用Ag2CO3作为氧化剂,通过Rh催化取代丙烯酸与炔烃的氧化偶联反应,从而获得α-吡喃酮类化合物。随后,Li和Rovis等[2,3]报道了使用化学计量的过渡金属氧化剂以及高温条件,实现了Rh催化丙烯酰胺和炔烃的C-H环化反应,从而获得α-吡啶酮(Fig. 1a, left side)。受此启发,已实现多种过渡金属催化(如Rh、Ru、Co、Pd、Fe等)炔烃的C-H环化反应,从而合成α-吡啶酮或α-吡喃酮。尽管已取得一定的成果,但仍存在一定的局限性:(1)较高的反应温度(100-120 ℃);(2)需化学计量的过渡金属氧化剂以再生催化剂,如Cu(OAc)2或AgOAc。
近年来,由于电流可替代传统的氧化和还原剂,电化学合成备受关注,已实现过电化学促进过渡金属催化(如Co,Ru,Rh和Cu)芳烃C-H与炔烃的环合反应。相比之下,用炔烃进行的电化学乙烯式C-H环化的研究较少。最近,作者课题组[4]报道了电化学Ir催化丙烯酸与内炔烃的乙烯基C-H环化反应,从而获得高收率的α-吡喃酮,但末端炔烃不适用于该体系。随后,Ackermann等[5]报道了在高温(140 ℃)条件下,通过电化学Ru催化实现丙烯酰胺与对称内炔烃的乙烯基C-H环化反应。在此,上海有机所梅天胜和浙江大学洪鑫课题组共同合作报道了电化学Rh催化丙烯酰胺与炔烃的C-H环化反应,从而获得良好至优异收率的α-吡啶酮和α-吡啶酮化合物(Fig. 1b)。同时,末端炔烃也作为良好的底物。
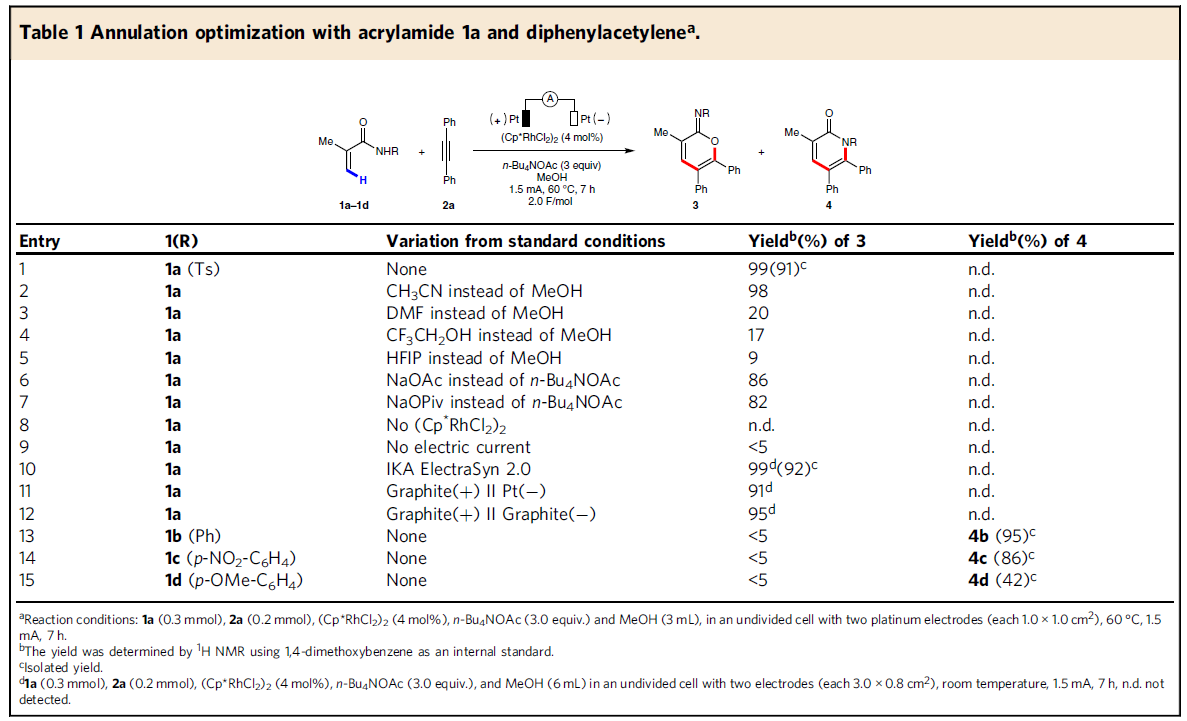
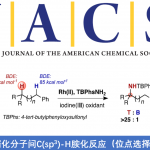
首先,作者以2-甲基丙烯酰胺(1a)和二苯基乙炔(2a)作为模型底物,进行了相关环化反应条件的筛选(Table 1)。反应的最佳条件为:以(Cp*RhCl2)2作为催化剂,使用n-Bu4NOAc为电解质,MeOH为溶剂,可在1.5 mA恒流电解条件下反应,获得91%收率的目标产物3a。
在获得上述最佳反应条件后,作者开始对底物进行了扩展(Fig. 2)。首先,各种烷基、酯、醚、芳基等取代的丙烯酰胺,均可顺利反应,获得相应的产物3a和6a–6r。其次,对于对称的二芳基乙炔和二烷基乙炔,均与体系兼容,获得相应的产物7a–7o。对于非对称的炔烃,则由芳烃电子性质控制区域选择性,如正丁基苯基乙炔(7p)可获得适度的区域选择性,而使用缺电子的芳基乙炔时(7q和7r),可以获得极佳的区域选择性。此外,一系列末端炔烃底物,均具有良好的区域选择性,获得产物7s–7x。值得注意的是,3a的克级实验同样取得预期的结果,进一步证明了反应的实用性(Fig. 2b)。

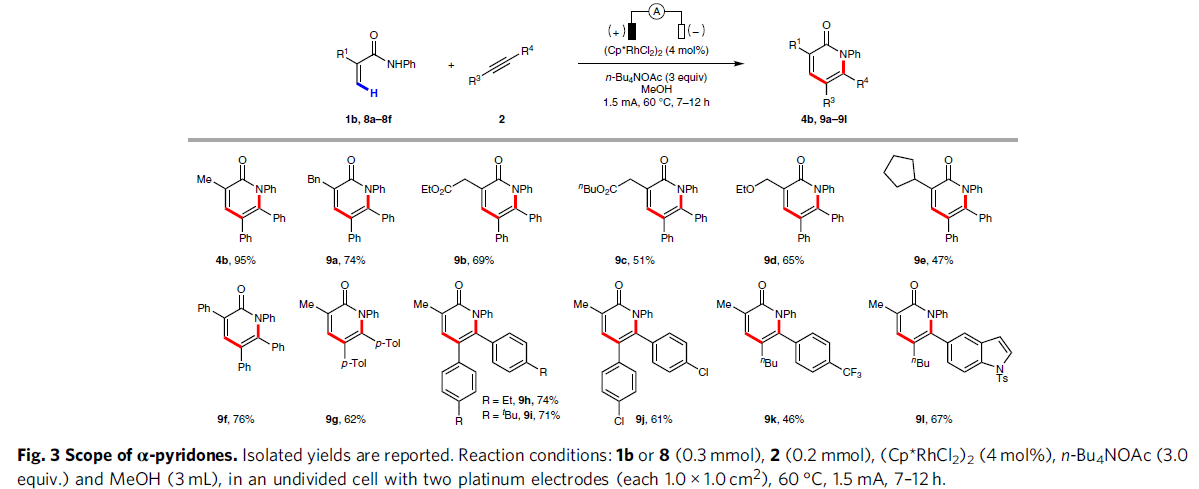
紧接着,在标准反应条件下,当使用一系列取代的丙烯酰胺,如烷基、酯、醚、芳基等,反应均具有良好的耐受性,从而以中等至良好收率的α-吡啶酮4b和9a–9l(Fig. 3)。
随后,作者开始对反应机理进行了进一步研究。首先,在无炔烃时,丙烯酰胺1a在标准条件下于CH3OD中反应时,观察到大量的H/D交换,表明C-H活化步骤是可逆的(Fig. 4a)。其次,通过比较丙烯酰胺5e和相应氘代底物5e–d4的平行实验,从而确定了动力学同位素效应(KIE)值为1.4(Fig. 4b)。

紧接着,在无电流时,将丙烯酰胺1a或5m、二苯乙炔2a置于标准条件下反应时,以高收率获得铑夹心配合物10和11(Fig. 5a)。同时,配合物10可在电化学条件下继续反应,获得产物3a(Fig. 5b)。此外,当以配合物10作为催化剂时,底物1a和2a可顺利反应,获得产物3a,从而表明配合物10作为电化学C-H环化中有效的中间体和催化剂(Fig. 5c)。

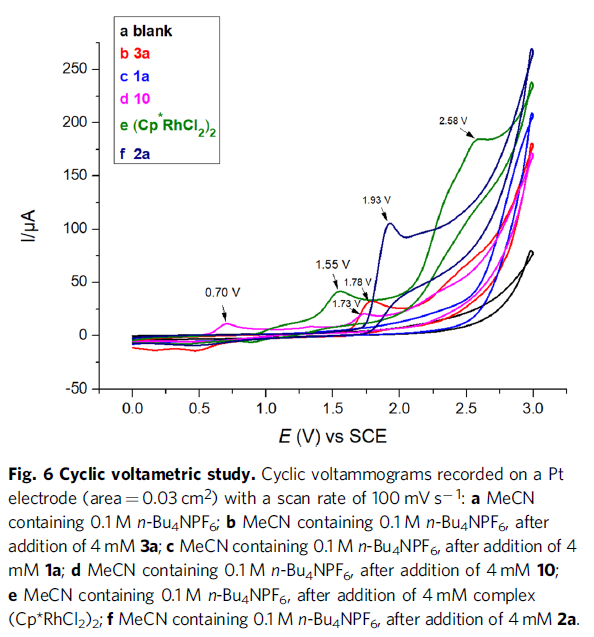
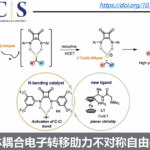
此外,通过循环伏安(CV)实验发现,配合物10在0.70 V处显示第一个氧化峰(曲线d),该峰明显低于反应中其他组分的氧化电位,从而表明阳极氧化作用是将二烯-Rh(I)配合物氧化为活性Rh(III)配合物,同时释放产物(Fig. 6)。
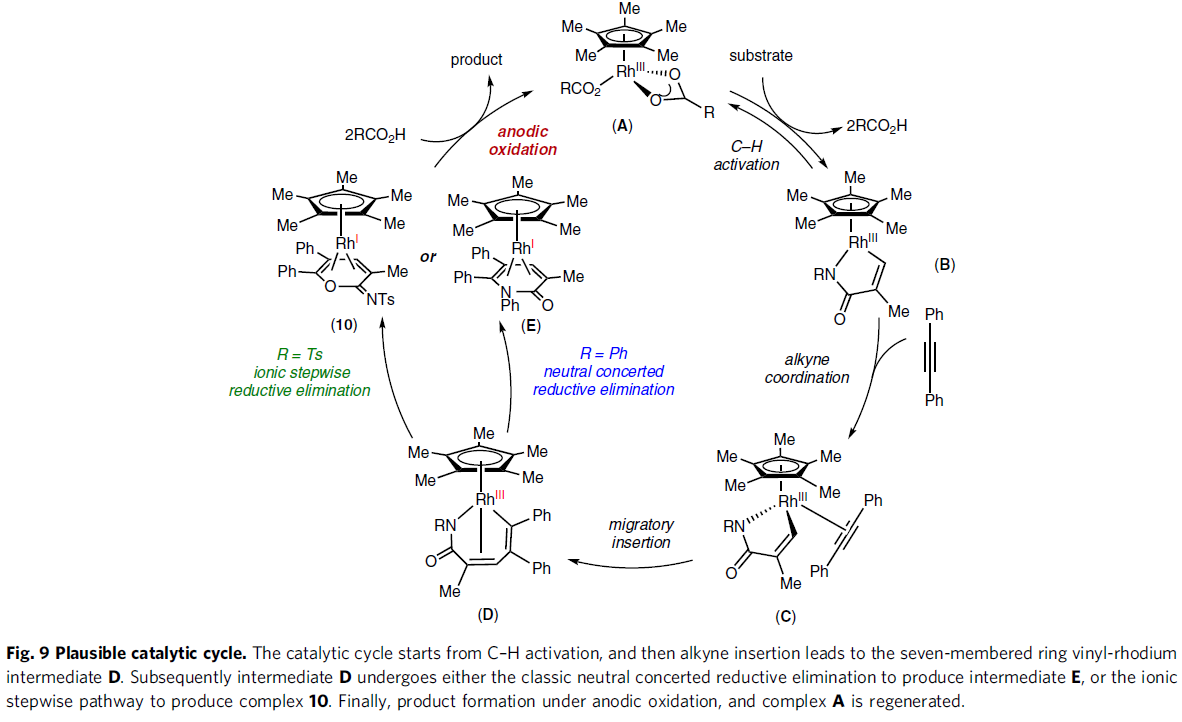
根据上述的实验,作者提出了一种可能的反应机理(Fig. 9)。首先,配合物A通过C-H活化形成环金属化的Rh(III)中间体B,再与炔烃配位获得配合物C。紧接着,配合物C经迁移插入形成七元铑配合物D,可经离子分步或中性协同还原消除,从而得到Rh(I)配合物10或配合物E。最后,中间体10或E经阳极氧化后,从而获得产物,并生成配合物A,已完成催化循环。
总结
上海有机所梅天胜和浙江大学洪鑫课题组共同合作报道了一种电化学Rh(III)催化丙烯酰胺与炔烃的C-H环化反应,从而获得良好至优异收率的α-吡啶酮和α-吡啶酮化合物。同时,当使用不对称炔烃时,反应具有极好的区域选择性。此外,反应涉及铑催化C-H活化和炔烃插入形成七元环乙烯基铑中间体,再经中性协同还原消除或通过离子分步途径以生成目标产物。
















No comments yet.