
本文作者:芃洋雪
导读
神奇的甲基效应(magic methyl effect)在药物化学中众所周知,然而对于药物分子的后期修仍具有一定的挑战。为了克服这一难题,瑞典阿斯利康公司(AstraZeneca)的Magnus J. Johansson和德国哥廷根大学的Lutz Ackermann教授在自然化学(Nature Chemistry)杂志上发表论文,通过钴催化剂,实现了对复杂药物分子后期的C-H甲基化反应。依赖于硼的甲基化试剂,同时利用分子内在的导向基团来活化C-H键,该反应具有广泛的适用性。此外,该反应无需预功能化或后脱保护,即可对各种市售药物分子和天然产物进行甲基化。同时,理化性质和生物学测试也证实了这种看似微小的结构变化,会影响重要的药物特性。
Cobalt-catalysed C–H methylation for late-stage drug diversification
Stig D. Friis, Magnus J. Johansson, Lutz Ackermann.
Nat. Chem. volume 12, 511–519 (2020),DOI:10.1038/s41557-020-0475-7
正文
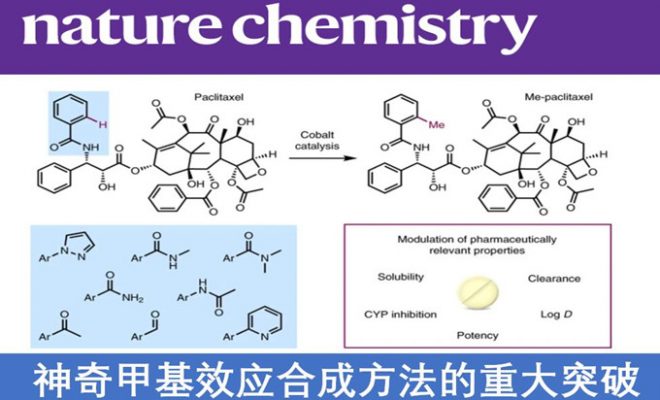
在药物开发中,后期官能团化或后期修饰至关重要。这种策略可直接对生物活性分子进行结构多样化修饰,从而避免从头合成,并迅速获得目标产物。过渡态金属催化的特定位置C-H活化,已实现在复杂分子上引入体积较小的官能团(如氟、氰基、胺或者短链烷烃)。而在药物结构中,C-H键上的氢被甲基取代后,有助于提高药物的结合亲和力、生物利用度、代谢稳定性,改变药物的药代动力学,从而被药物化学家称为“神奇的甲基效应”或者“魔幻的甲基效应”(magic methyl effect),如图1a所示。

图1a. 药物分子的C-H甲基化修饰. 图片来源:Nature Chem
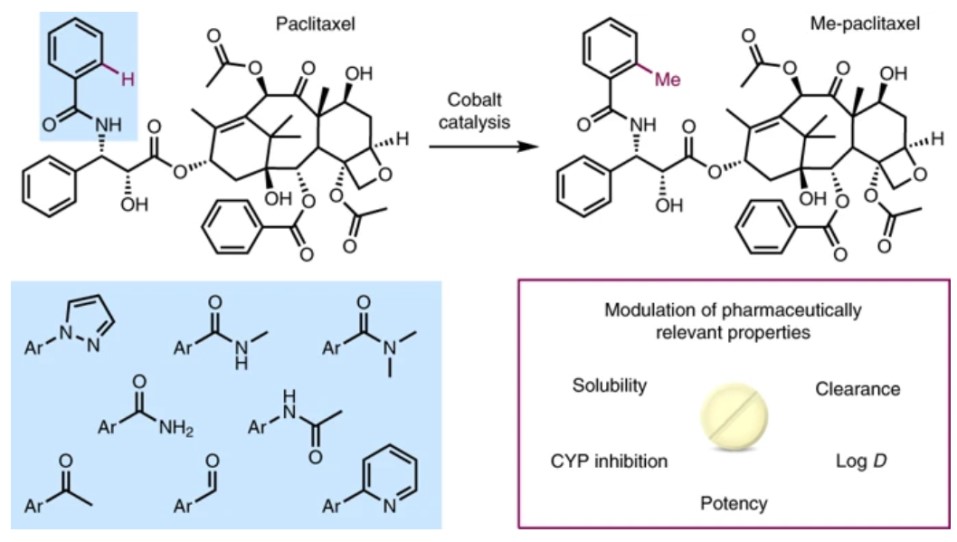
通过路易斯碱和过渡态金属的配位,可对分子结构特定位置上的C-H键直接活化,同时导向基团可以提高位置选择性和转化率。在C-H甲基化反应中,现有的导向基团一般需要偶联试剂(如格氏试剂),利用特殊的基于硼的烷基化试剂或者与贵重金属钯催化剂合用的阴离子导向基团(Fig. 1-b)。但在生物活性药物分子中,因结构复杂、官能团多,因此仍缺乏一种通用的合成方法。
图1b. 甲基化反应中的过渡态金属和甲基化试剂. 图片来源:Nature Chem
因此,作者提出以分子内的官能团作为导向基团,选用硼甲基化试剂,最关键的是以Cp*配位的(Cp* =C5Me5)高度亲电性的Co(Ⅲ)催化剂,可以和更弱的路易斯碱官能团配位。钴催化的甲基化反应与其他过渡态金属催化过程基本一致,首先钴催化剂和底物络合,经C-H键活化和金属交换反应后,甲基从硼试剂转移到钴上,再经还原消除,从而获得甲基化产物,同时Co(Ⅲ)经还原变为Co(I),再通过碳酸银氧化为Co(Ⅲ),从而完成催化循环(Fig. 1-c)。
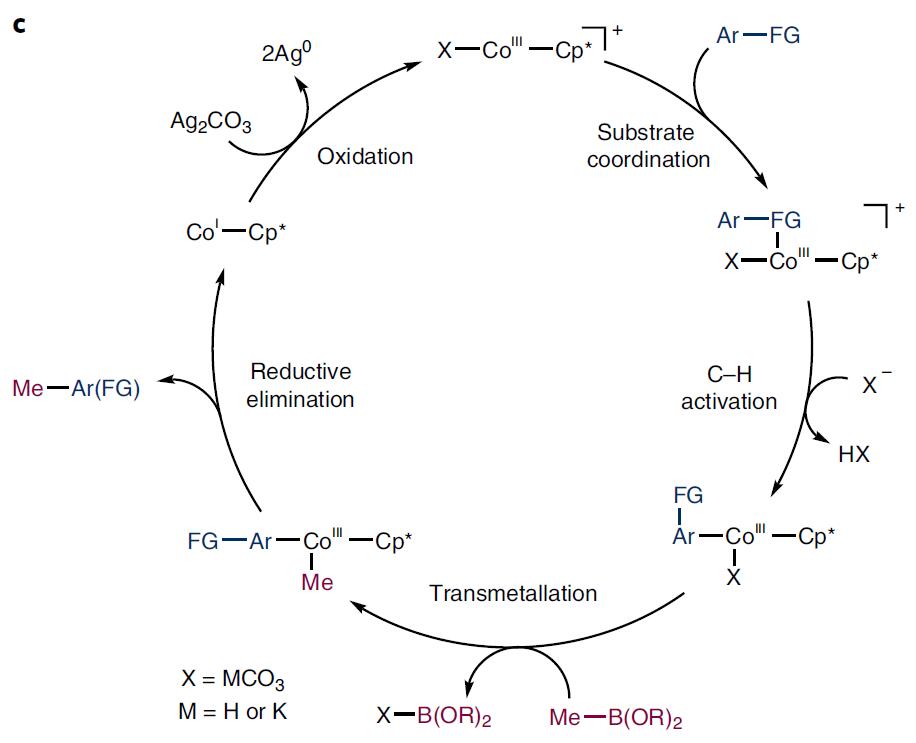
图1c. Co催化的甲基化反应机理. 图片来源:Nature Chem
目前钴催化的甲基化反应很少被研究,影响这类反应的参数极多,而且在有机合成中,难以对多种不同的参数定位,有时还存在两种参数相互关联的情况,如试剂的溶解度有限,在合适的有机溶剂中可能不溶解等问题。对此在合成方法条件优化中,作者采用了交叉参数优化(Cross-parameter optimization)的过程,从而将600百万个排列组合降至约2000个实验(图1d)。
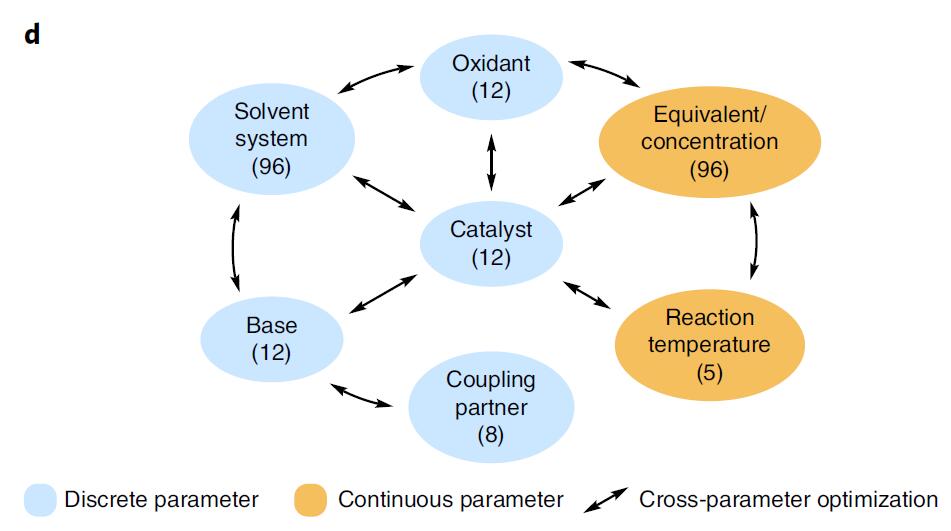
图1d. 反应条件优化的参数. 图片来源:Nature Chem
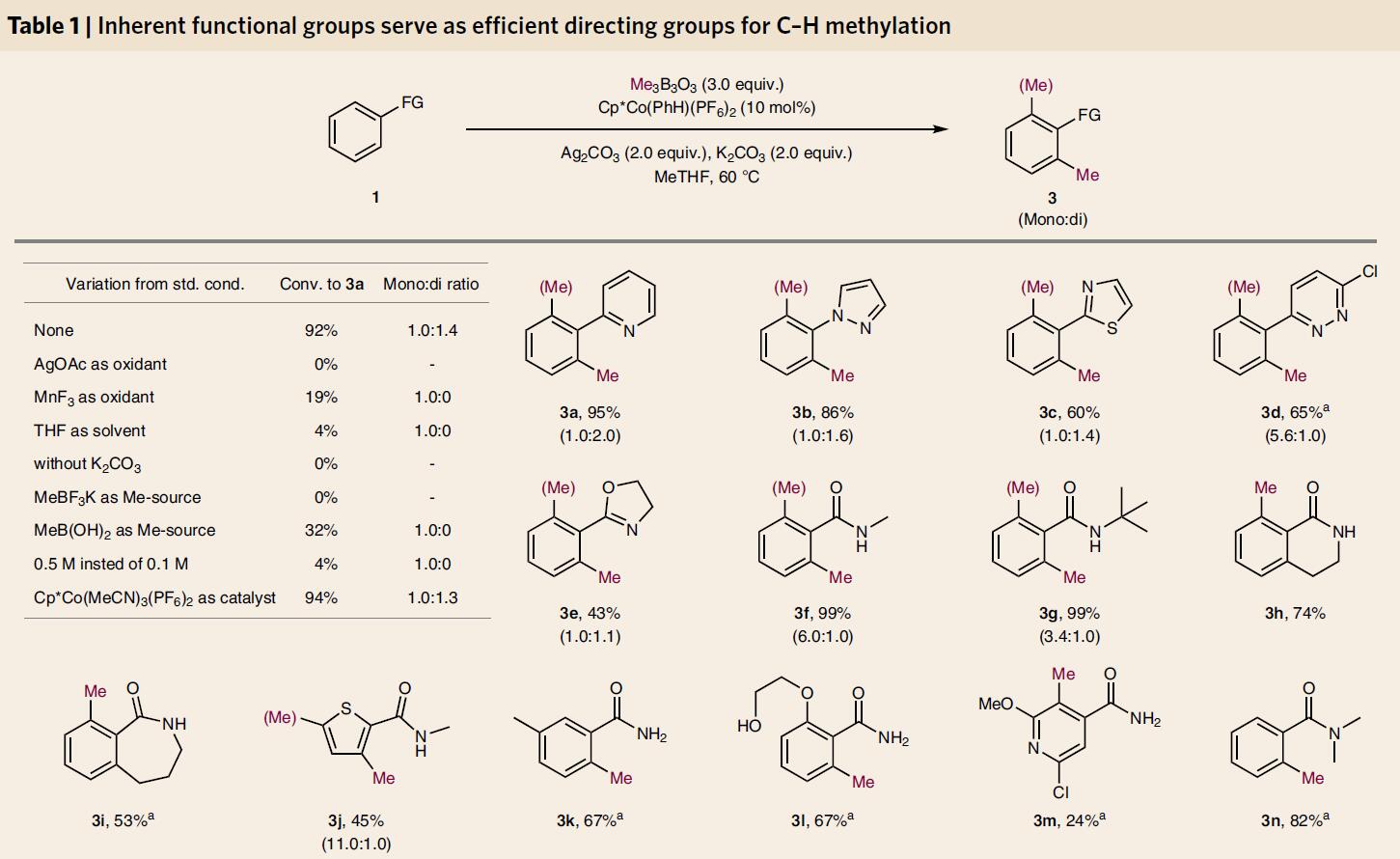
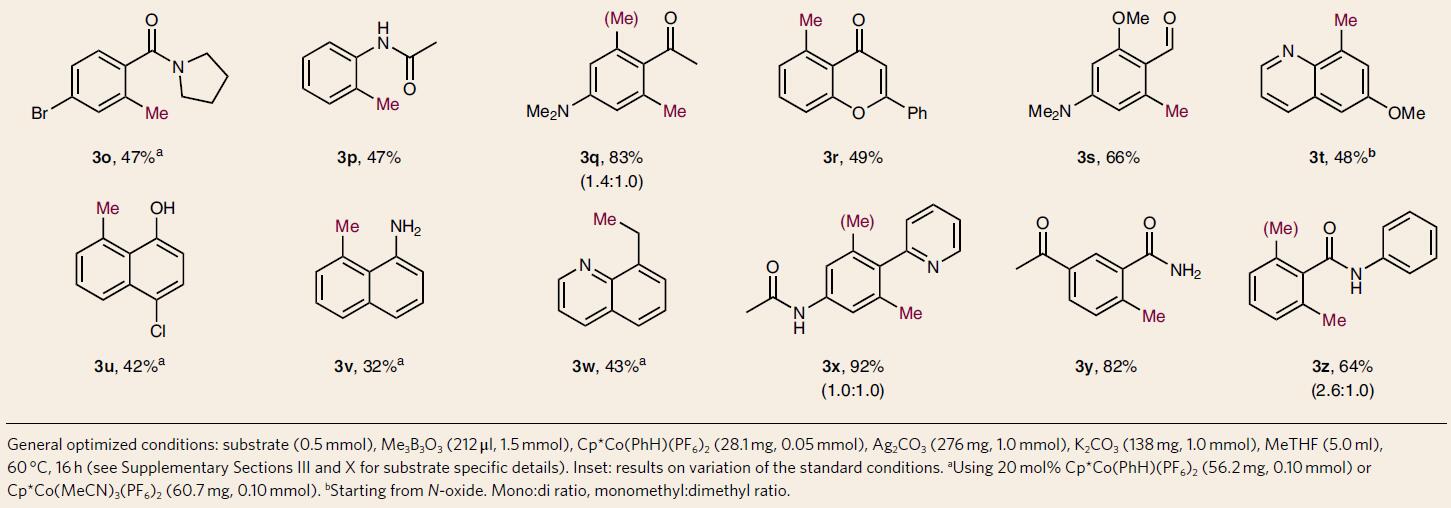
通过高通量实验筛选后,作者发现,当以2-甲基四氢呋喃为溶剂,Ag2CO3为氧化剂,三甲基环三硼氧烷为甲基化试剂,Cp*Co(PhH)(PF6)2作为催化剂,可获得最佳反应结果(Table 1)。紧接着,作者对底物范围进行了扩展(Table 1)。反应结果表明,药物分子中常见的氮杂环(3a–3e)或者酰胺基团(3f–3p),均与体系兼容。一级酰胺由于缺乏导向基团,从而导致具有合成难度,但增加催化剂用量,也可以实现邻位的甲基化(3k-3m)。二级苯甲酰胺(3f-3j)不论是否有立体位阻或环状,都是很好的底物。三级酰胺1n 和1o可以选择性的单甲基化,含溴的底物3o也没有出现副反应。底物3p和3z的产物出现了区域选择性产物。醛和酮与含氮相比碱性更弱,少见作为C-H活化的底物,但在富电子的芳香环上,这个方法可以生成3q和3s。稠环芳香烃,可以选择性的在1-位甲基化。然而,酯和羧酸不适用该体系。
同时,作者还进行了11种底物的竞争性反应,即2个底物为1组,看哪种底物可生成产物,根据产物生成情况将各种底物分为4组,分别为classⅠ高活性组,classⅡ混合活性组,classⅢ中等活性组和classⅣ低活性组,各种底物见图2所示。

图2. 底物的竞争性反应分组。图片来源:Nature Chem
在药物分子或天然产物中,结构复杂性以及官能团的敏感性,因此作者在反应中加入47种代表性官能团的化合物作为添加剂,通过288个实验研究兼容性,颜色为红色<25%,橙色25-50%,绿色>50%(图3)。结果表明,(1)DMSO可阻止反应,而水、乙腈、DMF等极性溶剂并不影响;(2)羧酸不参与反应,而醇和酚可以反应;(3)强配位作用的含氮杂环,如吡啶、N-甲基咪唑阻止了反应;(4)烯烃比炔烃更易反应。
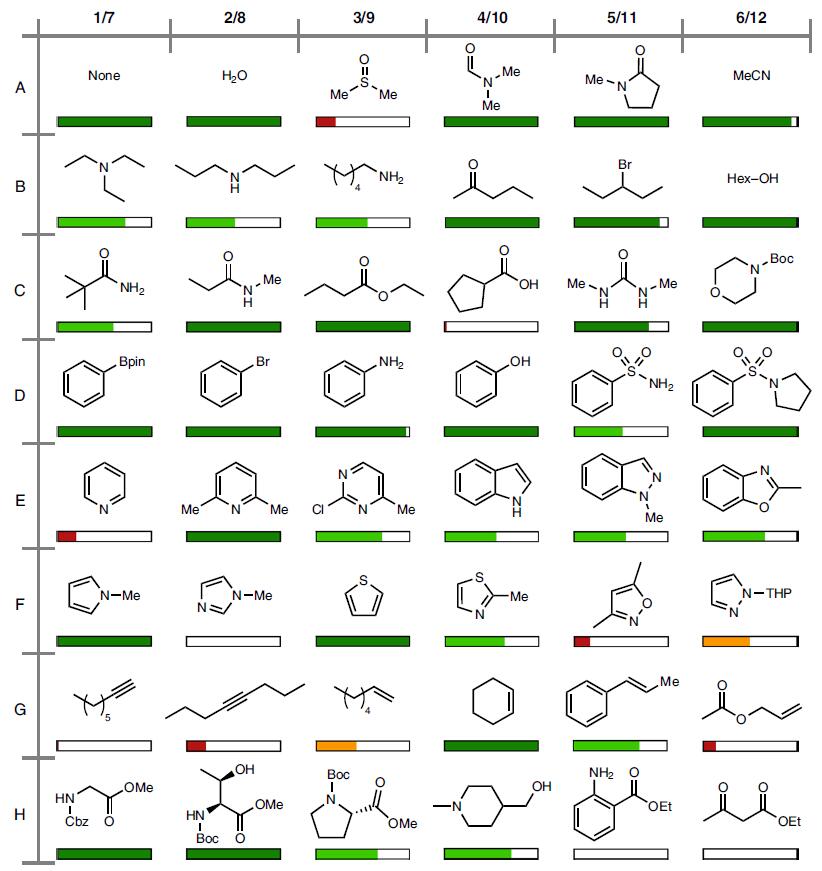
图3.考察添加物对反应的影响. 图片来源:Nature Chem
随后,作者对实用性进行了研究。由于天然产物具有结构复杂性、合成难度大的问题,从而导致对其结构修饰更大困难。对于药企而言,快速得到结构多样化的化合物更重要,若从头设计,会导致总收率低,同时造成时间和人力成本的浪费。对此,通过对天然产物和市售药物直接的甲基化修饰,可避免上述问题(Table 2)。用催化量的钴络合物4a对8个复杂、结构多样的化合物进行甲基化反应,得到12个甲基化的产物。而且,昂贵的未反应的底物方便回收。如降胆固醇药物非诺贝特(fenofibrate)得到两个异构体6d1和6d2(比例3:1)。紫杉醇(paclitaxel)有47个C-H键,14个氧原子和多个敏感官能团,可以生成甲基化产物6c,分离收率高达32%。塞来昔布(celecoxib)5i尽管有多个路易斯碱,增加催化剂4a的用量至化学量经HPLC纯化后,可获得总收率为30%的6i1和6i2。氟哌啶醇 (haloperidol)也获得较高收率的6j。依伐卡托(ivacaftor)的甲基化产物有3个(6m1,6m2,6m3),总收率仅有28%。对此,作者列举了各种复杂药物分子的结构以及文献报道的合成步骤,而且仅有<15%有甲基化修饰产物,显然后期甲基化修饰具有绝对优势。
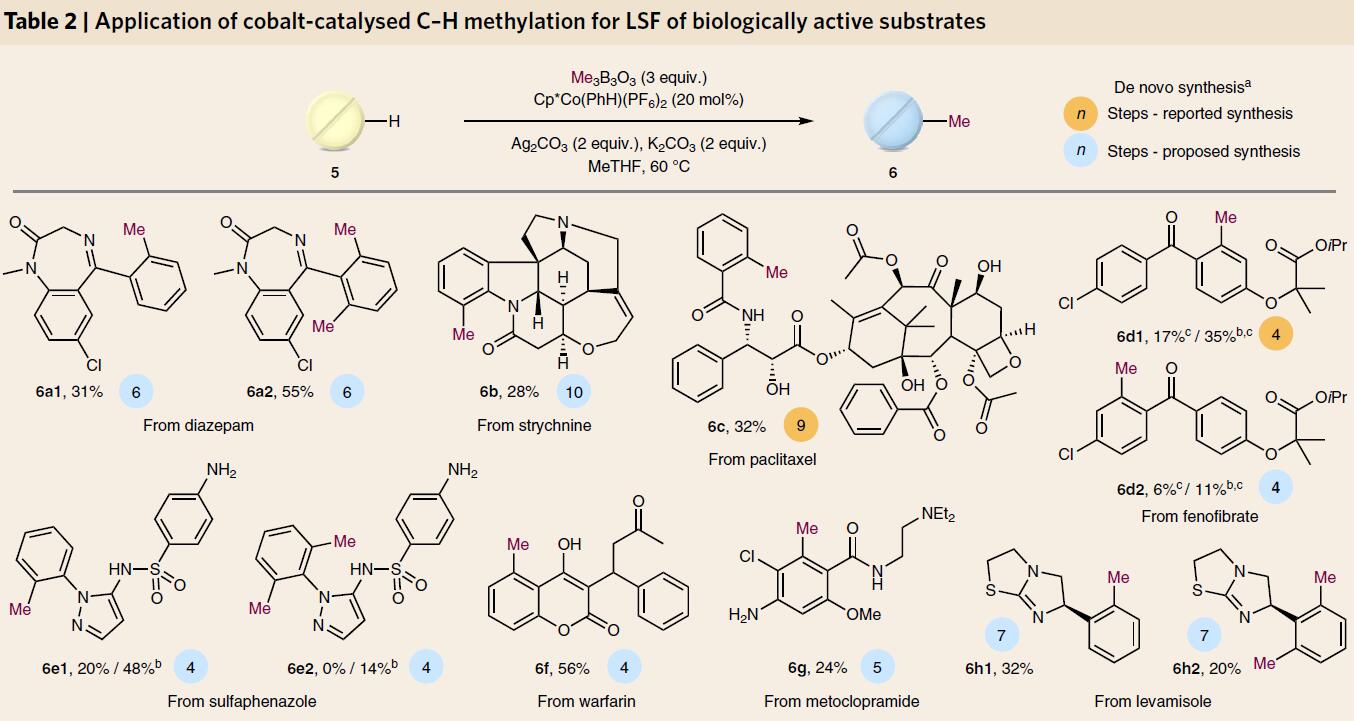
表2 生物活性分子的甲基化修饰


图4. 生物活性分子的甲基化修饰。蓝色圈里的数字表示文献报道的合成步骤,
橙色圈为预测的合成步骤. 图片来源:Nature Chem
调节药学特性
在获得上述的甲基化产物后,作者开始对药效学和药物代谢及药代动力学DMPK进行了研究(图5)。
LogD
很多DMPK特征与log D密切相关,但甲基化药物分子的log D并不是全部增加,如甲氧氯普胺(metoclopramide)5g和阿普斯特(apremilast)5k,见图5a,浅蓝色为底物,深蓝色为产物。
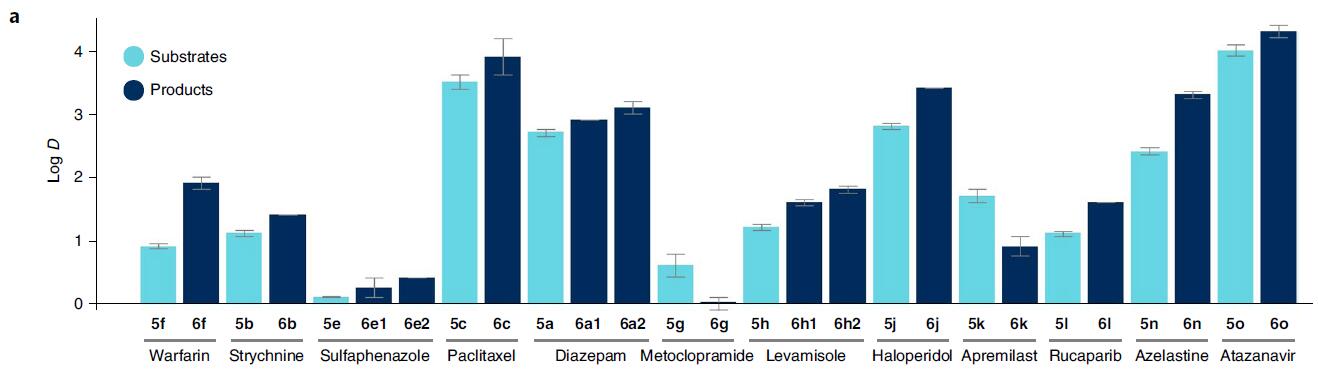
图5a. 底物及甲基化产物的logD图. 图片来源:Nature Chem
代谢稳定性
紫杉醇甲基化后,增加了溶解性,并且减少了在老鼠肝细胞和人肝微粒体中的固有清除率(CLint),增加了代谢稳定性。虽然苄基甲基被认为是对细胞色素P450酶易于氧化的基团,但在甲基化修饰的药物中反而增加了代谢稳定性,尤其是甲基化的氟哌啶醇(haloperidol)5jvs6j。
血浆蛋白结合率
甲基化修饰后,还可以改变药物的血浆蛋白结合率,如游离的安定(diazepam)5a(2.0%)vs甲基化的安定6a1(8.5%)和6a2(9.0%),氮卓斯汀(azelastine)5n(15%)vs甲基化产物6n(4.6%)。
CYP酶抑制性
避免或减少CYP酶的降解,是优化先导化合物重要指标,而甲基化则是个重要的修饰方法。人体内代谢药物的主要酶是细胞色素P450超家族(Cytochrome P450 proteins, CYP),有些药物甲基化后可以抑制,有些则不能,这和甲基化的位置相关。甲基化是改变CYP酶抑制性的重要方法,但现在不能预测是否能够抑制酶的活性。
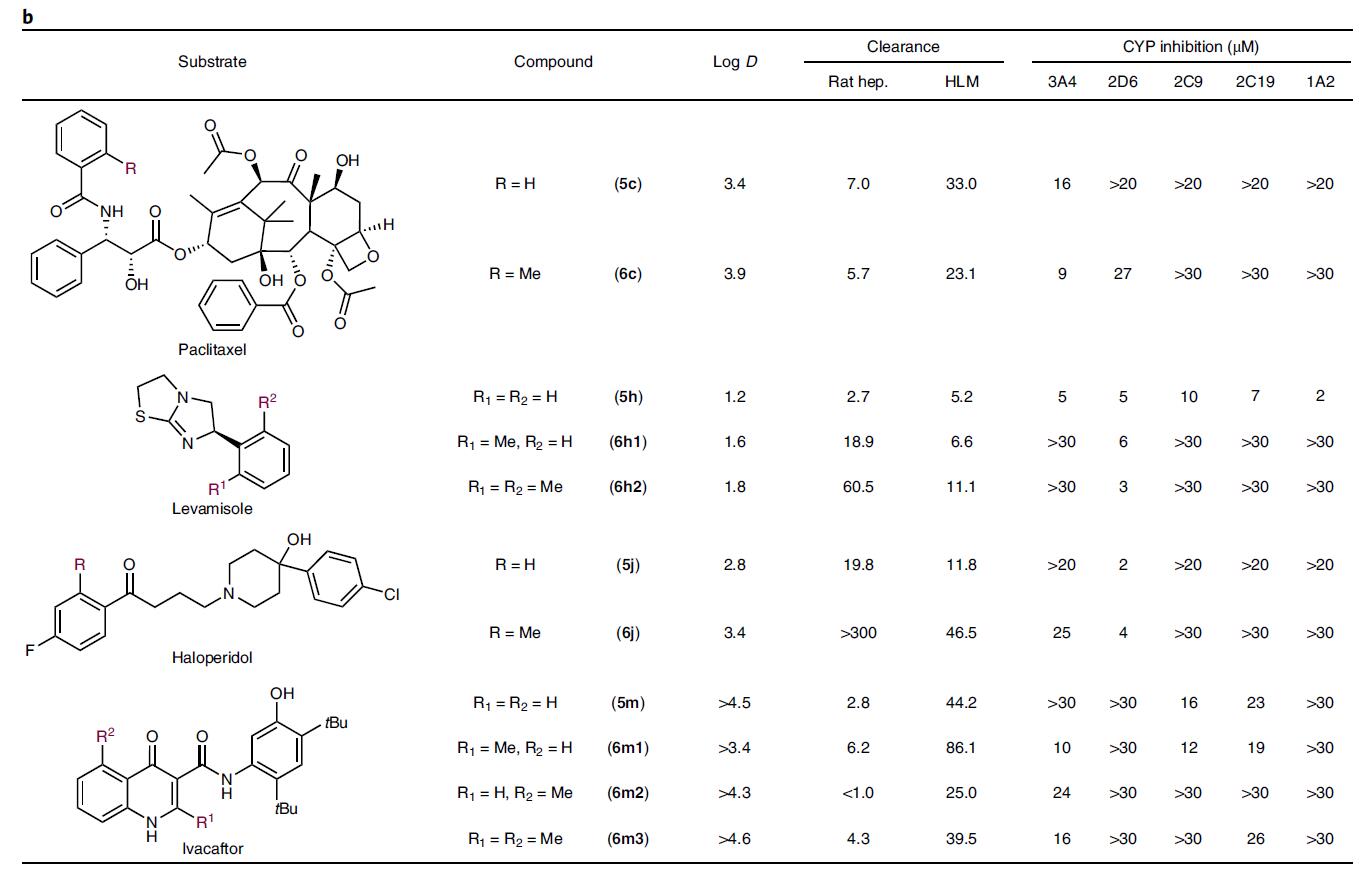
图5b. 底物及甲基化产物的药效学数据. 图片来源:Nature Chem
总结
来自阿斯利康和哥廷根大学的工作者,不仅开发了一种通用的后期甲基化的合成方法,还详细介绍了交叉参数优化的过程,从而将600百万个排列组合降至约2000个实验。作者先筛选了不同官能团的竞争性和兼容性后,再进行上市药物甲基化反应,并且对甲基化的产物进行了详细的药效学研究,展现了神奇的甲基化效应,又因不能完全预测又具有魔幻性。全文内容详实,研究既有深度又有广度,不仅如此,全文配图极具美观,颇有可读性。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!
















No comments yet.