
本文作者:ChemBoy
导读
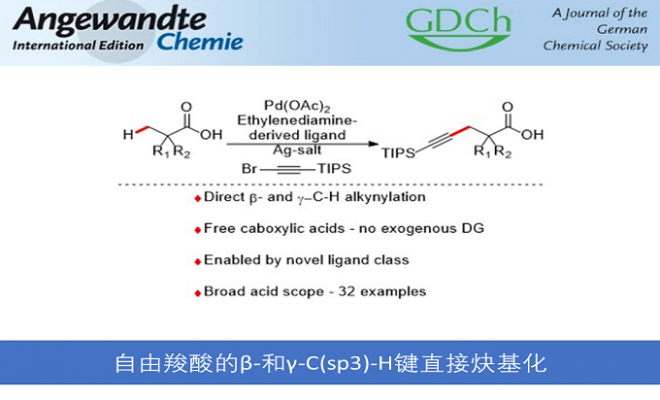
近日,德国明斯特大学Gemmeren教授课题组成功实现了一种新型配体,可促进钯催化的自由羧酸类底物的β-和γ-位C(sp3)-H键直接炔基化反应。该方法能避免导向基的引入与脱除的步骤,并且底物范围广泛,不仅适用于α-位季碳中心类羧酸底物,还适用于α-位非季碳中心类羧酸底物。此外,作者还初步发展了该反应的对映选择性版本。相关成果发表在《Angew. Chem. Int. Ed.》上:
“Direct β-andγ-C(sp3)–H Alkynylation of Free Carboxylic Acids”
Francesca Ghiringhelli, Alexander Uttry, Kiron Kumar Ghosh, Manuel van Gemmeren*
Angew. Chem. Int. Ed. 2020, Just Accepted, DOI: 10.1002/anie.202010784
正文
前言
炔烃已被广泛应用于药物化学、材料科学与合成化学中。由于炔基具有合成的多功能性,常作为合成过程中关键的部分,可通过后续的转化从而合成各种新材料与药物分子。因此,有必要发展高效的炔基引入的方法。在该领域,C-H键的直接炔基化反应已受到了广泛关注[1]。
羧酸原料丰富、廉价易得,且炔基化获得的产物含有两个过高度多功能性官能团,因此,过渡金属催化的C(sp3)-H键直接炔基化反应备受关注。在这方面,直接利用非官能化的自由羧酸作为底物实现其C(sp3)-H键活化/炔基化,作为一种理想的方法(Scheme 1, Path A)。然而,这种方法的发展受到底物固有羧基作为导向基的阻碍,由于羧基自身的弱导向能力以及羧基与催化剂钯中心之间存在竞争配位模式[2],因而该方法在合成上具有很大的挑战。已报道的C-H键炔基化反应中,通过引入外加导向基可克服这些挑战,但是这增加了整个转化过程中的合成步骤(Scheme 1, Path B)[3]。2011年,Chatani等人利用双齿导向基首次实现了钯催化脂肪羧酸的惰性C(sp3)-H键直接炔基化[4]; 随后,Yu课题组发展了含多氟芳基脂肪酰胺的β-甲基C(sp3)-H键的炔基化[5], 在2017年,该课题组报道了第一例C(sp3)-H键对映选择性炔基化[6],以及第一例寡肽C(sp3)-H键炔基化反应[7]。同时,其他课题组也发展各种新的催化体系与导向基成功实现了C(sp3)-H键炔基化, 如Chen, Shi, Colobert及Verho课题组[8]。此外,其他钯催化的炔基化及双齿导向基导向钴和镍催化的反应已被报道[9]。尽管上述所报道的文献中的方法均能获得目标产物,但是这些方法仍离理想的原子与步骤经济性反应还较远。最近,作者课题组及其他课题组已经证实通过合理的配体设计,能够在短周期内实现自由羧酸的惰性C(sp3)-H键活化/官能团化,如芳基化[10]、乙酰氧基化[11]、内酯化[12]及烯基化反应[13]。
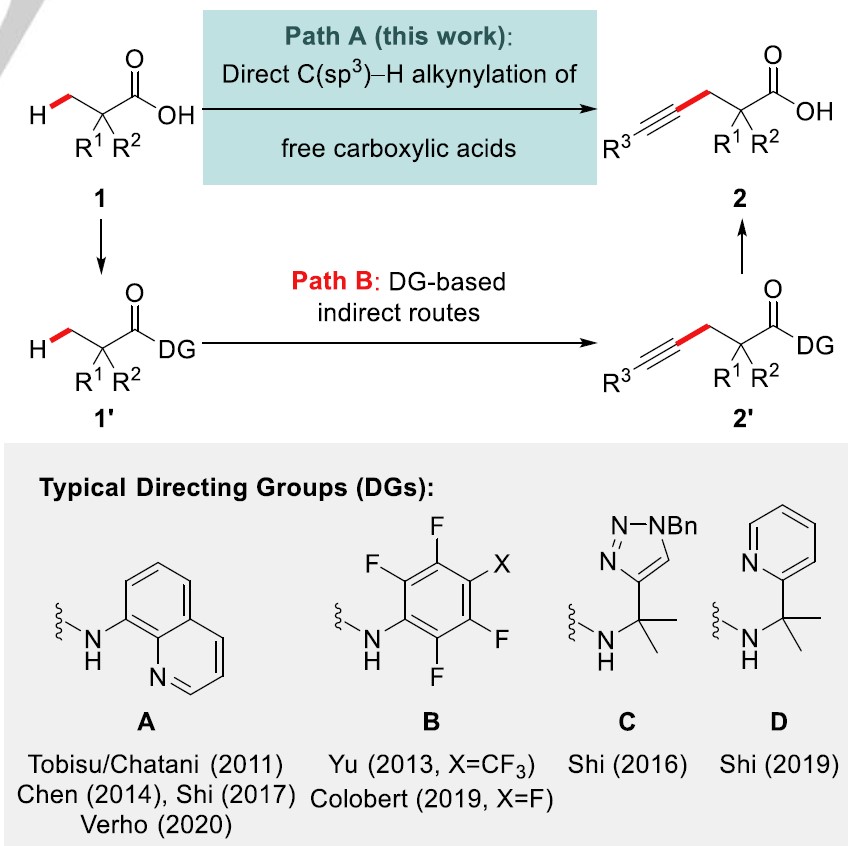
Scheme 1. Previous studies on the indirect C−H alkynylation of aliphatic acids (Path B) and direct C(sp3)−H alkynylation of free carboxylic acids developed in this study (Path A).
反应条件优化
作者基于本课题组在自由羧酸的β-和γ-位C(sp3)-H键直接官能团化的研究经验,认为合适的配体对实现自由羧酸C(sp3)-H键的炔基化反应至关重要。作者以特戊酸1a为模板底物、三异丙基硅基炔基溴为炔基化试剂,Pd(OAc)2为催化剂、Ag2O作为氧化剂、LiHFIP作为碱、HFIP为反应溶剂对配体进行筛选(Scheme 2)。作者发现在自由羧酸C(sp3)-H键的其他类型转化中使用的配体,氨基酸类配体L1-L3、硫醚类配体L4、单保护的胺基乙胺配体L5、邻氨基苯甲酸类配体L6及噁唑啉配体L7均不能获得令人满意的结果。
随后,作者发现一种新型二胺类衍生物配体能有效促进该反应的进行。当使用配体L8时,能以10%的产率生成目标炔基化产物,接着继续对配体进行进行筛选,发现芳基取代的磺酰胺配体L9能够明显促进该反应的进行,产率明显升高(65%)。接着,作者继续对芳基上含不同取代基的磺酰胺类配体进行测试(L10-L13),发现对产率没有明显的影响;最后,作者对乙二胺骨架上含不同芳基类配体进行筛选(L14-L18),发现对反应的产率也没有明显提高。由于配体L9结构相对较简单且易获得,故选择L9为配体进一步研究。
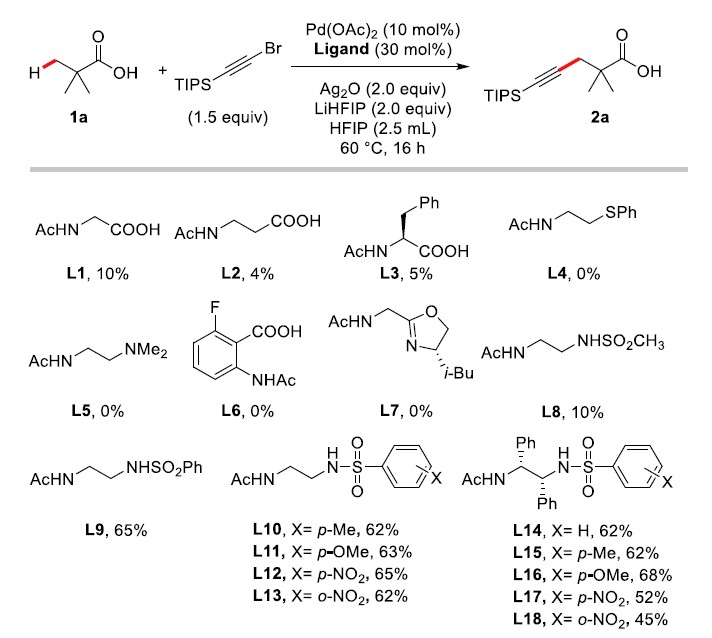
Scheme 2. Identification of suitable ligands for the β- and γ-C(sp3)–H alkynylation of free carboxylic acids.
底物拓展
在确定最优反应条件后,作者对该反应的底物普适性进行研究(Scheme 3)。在标准反应条件下,特戊酸1a以65%的产率生成目标产物2a;对底物脂肪链进行改变,也能以良好的产率获得目标产物2b(70%)、2c(63%)和2d(65%);脂肪链末端氟取代(2e, 57%)、TBSO-取代(2f, 55%)、-OH取代(2g, 45%)均能能顺利进行反应。作者进一步对含不同芳基取代的自由羧酸类底物进行评估,发现该反应具有很好的官能团兼容性,无论芳环上给电子基取代(2i, 2j)还是吸电子基取代(2k-2r)类底物在该反应条件下均能兼容。有趣的是,苯基上无其他取代基底物(2h, 58%)和萘环取代的底物(2s, 59%)均能以良好的产率生成目标炔基化产物,并且芳基取代的底物在该反应条件下未观察C(sp2)-H键炔基化竞争反应的发生。重要的是,一些与生物活性相关的官能团也能很好的兼容,如醚(2j, 63%)、氰基(2k, 57%)、三氟甲基(2l, 58%)、三氟甲硫基(2m, 55%)、酯基(2n, 57%)、磺酸酯基(2o, 68%)、硝基(2p, 75%)和酮羰基(2q, 67%)。此外,在该反应条件下能实现环丙烷羧酸底物的β-位亚甲基C(sp3)-H键的炔基化反应(2t, 45%, 98:2 dr)。
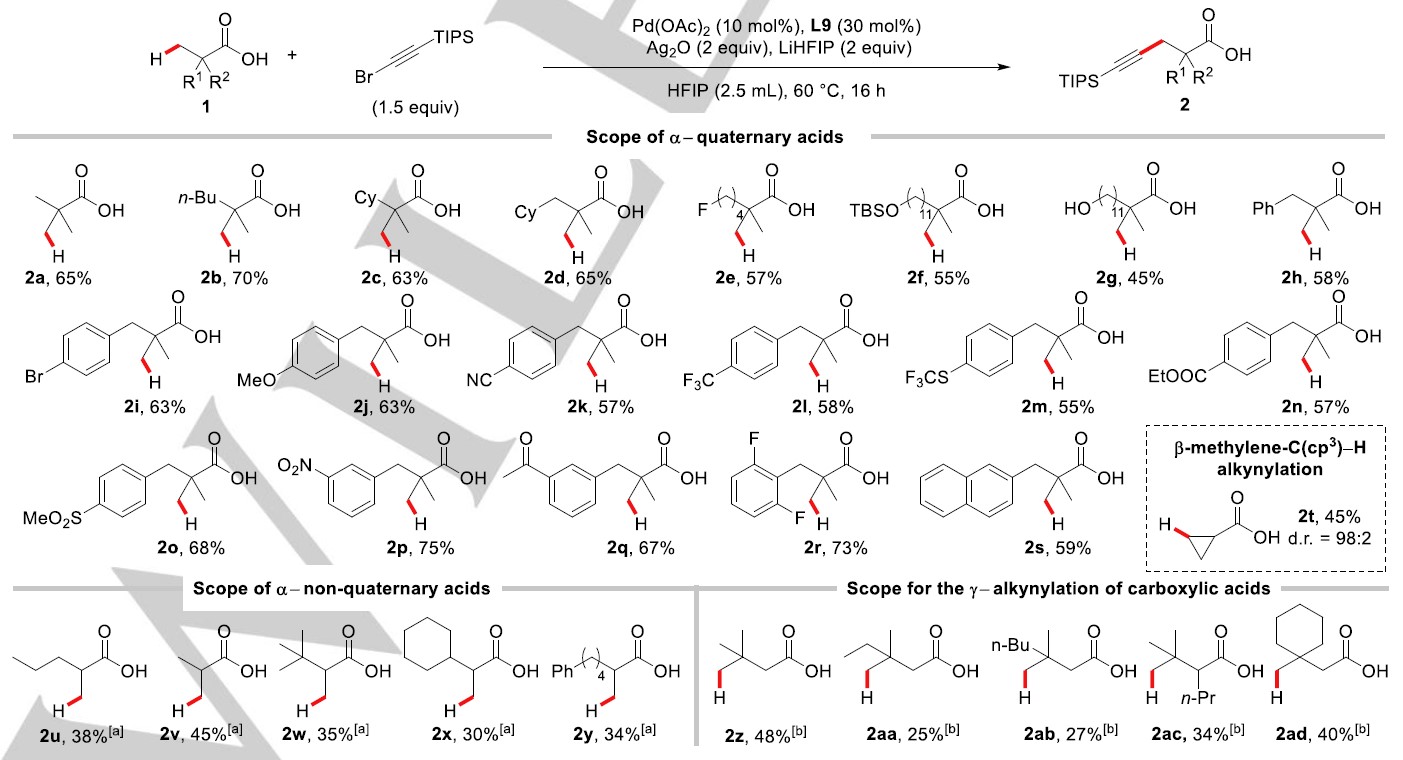
Scheme 3. Reaction scope. [a] L14 was used instead of L9, LiHFIP (1 equiv), H2O (1.5 equiv), and HFIP (2.0 mL) were used at 60 °C for 24 h; [b] Pd(OAc)2 (20 mol%), L14 (60 mol%), LiHFIP (2 equiv), Ag2O (2 equiv), 1-bromo-2-(triisopropylsilyl)acetylene (3 equiv), and HFIP (4 equiv) were used at 60 °C for 24 h.
为了进一步拓宽该反应方法的应用范围,作者对极富挑战性的α-位是非季碳中心的脂肪羧酸类底物进行了探索。在许多研究报道中,发现这类脂肪羧酸底物在其他类型的C-H键活化/官能团化反应中具有非常大的挑战性,这是由于这类底物不存在偕二甲基效应的促进作用以及可能存在其他副反应。令人高兴的是,作者对反应条件进行调整,将配体L9换成L14、碱减少至1.0当量、加入1.5当量H2O、溶剂HFIP减少至2.0 mL、反应时间延长至24h,发现这类底物能够顺利进行目标C-H键炔基化反应(2u-2y)。值得注意的是,尽管这类底物利用该反应方法获得的炔基化产物产率不高,但避免了外加导向基团的引入与脱除,有效提高了效率。
基于这种配体促进的自由羧酸的β-位C(sp3)-H键活化策略以及以经报道了更具挑战性的远程γ-位C(sp3)-H键直接官能团化转化,作者进一步将对自由羧酸的γ-位C(sp3)-H键炔基化反应进行研究。作者在标准反应条件基础上进行调整,催化剂负载量增加至20 mol%、配体L14增至60 mol%、炔基化试剂增加至3.0 当量、溶剂减少至4.0当量,反应24h,能以中等产率生成γ- C(sp3)-H键炔基化产物(2z-2ad)。该方法获得的目标产物产率虽然不高,与已经报道的利用外加导向基实现的炔基化反应相比产率较好一些;此外,该方法能够避免外加导向基引入与脱除过程中原料和产物的损失,这进一步增加了该反应方法的价值。
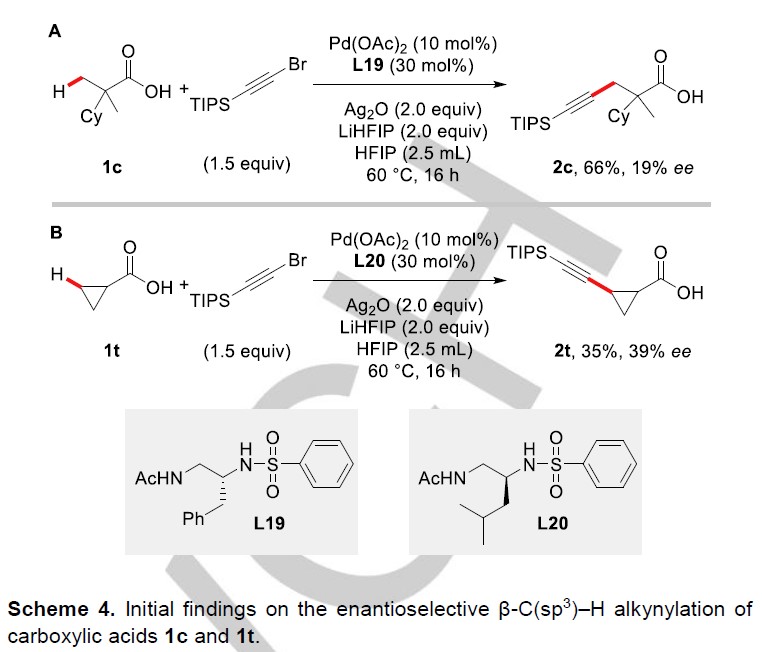
在对底物范围的研究结束后,作者该反应的对映选择性版本进行了探索。作者发现,在配体L9骨架上引入苄基后的配体L19能够对1c进行去对称化,以66%的产率及19% ee获得炔基化产物2c(Scheme 4a);类似地,将苄基换成异丁基后的配体(L20)能使环丙烷羧酸1t发生去对称化,以35%的产率及39% ee生成目标产物2t(Scheme 4b)。
为了证明该反应方法的实用性,作者对模板底物1a进行了大量反应实验以及对生成的产物2a进行后期衍生化应用实验(Scheme 5)。首先,作者进行5 mmol底物规模的放大反应,能以60%的产率生成目标产物2a(Scheme 5A);随后,作者对产物2a进行脱保护、接着进行Sonogashira偶联反应能以两步42%的产率生成化合物3,同时,作者先对2a进行酯化、再脱保护,接着进行Sonogashira偶联反应能以三步67%的产率获得化合物4(Scheme 5B)。

Scheme 5. Synthetic utility of the β-C(sp3)–H alkynylation of aliphatic carboxylic acids.
机理研究
最后,作者该反应的机理进行了初步探索。首先,作者通过竞争实验和平行实验测定了KIE(Scheme 6A, KIE(parallel)=3.5、KIE(competition)=6.0),一级动力学同位素效应表明C-H键活化步骤是反应过程的决速步骤;紧接着,作者对C-H键活化步的可逆性进行了评估,分别在有炔基化试剂与无炔基化试剂的条件下反应(Scheme 6B),通过对剩余原料中的氘代比例进行分析发现,在无炔基化试剂存在条件下原料被高度去氘化,表明该反应中C-H键的活化步原则上是可逆过程,然而在有炔基化试剂存在条件下底物1b未发生去氘化。
根据这些观察到的结果,作者总结出该反应过程经历了C-H键活化决速步骤,虽然原则上C-H活化步是可逆的,但该炔基化反应观察到是不可逆的转化。根据已经报道的文献[1, 14],作者认为该反应的后续催化循环存在两种可能:一种是Pd(IV)/Pd(II)路径,即C-Br键与C-H活化后形成的金属中间体发生氧化加成,接着发生还原消除形成C-C键;另一种可能是Pd(II)路径,即炔烃与C-H活化后形成的金属中间体发生迁移插入,接着发生Pd-Br消除。在这两种可能的路径中,银盐均可起到活化炔烃与结合Br-以避免催化剂中毒的作用。最后,LiHFIP作为碱可以避免反应过程中除溶剂以外的其他共轭酸分子的生成,同时碱金属离子的存在才可以诱导羧基以κ1的配位模式与钯金属中心进行配位,从而促进C-H键活化步骤的发生[11a]。

Scheme 6. Preliminary mechanistic studies.
总结与评价
德国明斯特大学Gemmeren教授课题组开发了一种新型自由羧酸β-和γ-位C(sp3)-H键直接炔基化反应方法,新型乙二胺骨架配体的使用对于反应至关重要。该反具有底物范围广、官能团兼容性高等优点,不仅适用于α-位季碳中心类底物,还适用于α-位非季碳中心类自由羧酸的目标C-H键直接炔基化反应。同时,该反应避免了导向基团的引入与脱除步骤,进一步体现了步骤的经济性;此外,作者还初步发展了该反应的不对称炔基化反应以及对反应实用性进行了研究。最后,作者对反应机理进行了初步探索。
(注:本文章中所有图片均来自Angew. Chem. Int. Ed. 2020, Just Accepted, DOI:10.1002/anie.202010784)
参考文献
- [1] J. P. Brand, J. Waser, Chem. Soc. Rev. 2012, 41, 4165. DOI:10.1039/c2cs35034c.
- [2]Uttry, M. van Gemmeren, Synthesis, 2020, 52, 479. DOI: 10.1055/s-0039-1690720.
- [3] a) K. M. Engle, T. S. Mei, M. Wasa, J. Q. Yu, Acc. Chem. Res. 2012, 45, 788. DOI:10.1021/ar200185g;b) C. Sambiagio, D. Schönbauer,R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes, M. Schnürch, Chem. Soc. Rev. 2018, 47, 6603. DOI:10.1039/c8cs00201k.
- [4] Y. Ano, M. Tobisu, N. Chatani, J. Am. Chem. Soc. 2011, 133, 12984. DOI: 10.1021/ja206002m.
- [5] a) J. He, M. Wasa, K. S. L. Chan, J. Q. Yu, J. Am. Chem. Soc. 2013, 135, 3387. DOI:10.1021/ja400648w;b) H. Fu, P.-X. Shen, J. He, F. Zhang, S. Li, P. Wang, T. Liu, J.-Q. Yu, Angew. Chem. Int. Ed. 2017, 56, 1873. DOI:10.1002/anie.201610426.
- [6] Q. F. Wu, P. X. Shen, J. He, X. B. Wang, F. Zhang, Q. Shao, R. Y. Zhu, C. Mapelli, J. X. Qiao, M. A. Poss, J. Q. Yu, Science. 2017, 355, 499.DOI: 10.1126/science.aal5175.
- [7] T. Liu, J. X. Qiao, M. A. Poss, J.-Q. Yu, Angew. Chem. Int. Ed. 2017, 56, 10924. DOI:10.1002/anie.201706367.
- [8] a) B. Wang, C. Lu, S. Y. Zhang, G. He, W. A. Nack, G. Chen, Org. Lett. 2014, 16, 6260. DOI:10.1021/ol503248f; b) Y. Q. Han, Y. Ding, T. Zhou, S. Y. Yan, H. Song, B. F. Shi, J. Am. Chem. Soc. 2019, 141, 4558. DOI:10.1021/jacs.9b01124;c) S. Jerhaoui, J. P. Djukic, J. Wencel-Delord, F. Colobert, ACS Catal. 2019, 9, 2532. DOI: 10.1021/acscatal.8b04946;d) A. Balliu, A. R. F. Strijker, M. Oschmann, M. Pourghasemi Lati, O. Verho, 2020, DOI:10.26434/CHEMRXIV. 12034743.V1.
- [9] J. Zhang, H. Chen, C. Lin, Z. Liu, C. Wang, Y. Zhang, J. Am. Chem.Soc. 2015, 137, 12990.DOI:10.1021/jacs.5b07424
- [10] L. Hu, P. Shen, Q. Shao, K. Hong, J. X. Qiao,J. Yu, Angew. Chem. Int. Ed. 2019, 58, 2134. DOI:10.1002/anie.201813055.
- [11]a) K. K. Ghosh, A. Uttry, A. Koldemir, M. Ong, M. van Gemmeren, Org. Lett. 2019, 21, 7154DOI:10.1021/acs.orglett.9b02741;b) Z. Zhuang, A. N. Herron, Z. Fan, J. Q. Yu, J. Am. Chem. Soc. 2020, 142, 6769. DOI:10.1021/jacs.0c01214.
- [12] Z. Zhuang, J. Q. Yu, Nature, 2020, 577, 656.DOI:10.1038/s41586-019-1859-y.
- [13] a) Z. Zhuang, C. Bin Yu, G. Chen, Q. F. Wu, Y. Hsiao, C. L. Joe, J. X. Qiao, M. A. Poss, J. Q. Yu, J. Am. Chem. Soc. 2018, 140, 10363. DOI:10.1021/jacs.8b06527; b) K.K. Ghosh, A. Uttry, A. Mondal, F. Ghiringhelli, P. Wedi, M. van Gemmeren, Angew. Chem. Int. Ed. 2020, 59, 12848. DOI:10.1002/anie.202002362.
- [14] A.Mondal, H.Chen, L. Flämig, P. Wedi, M. van Gemmeren, J. Am. Chem. Soc. 2019, 141, 18662.DOI:10.1021/jacs.9b10868.
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!













No comments yet.