
本文作者:杉杉
导读
近日,南开大学叶萌春教授课题组在德国应化杂志发表论文,报道了在镍催化下(手性铝的控制),通过C-CN键活化,实现芳基腈和炔烃的对映选择性的多米诺反应,反应可在温和的条件下以32-91%的收率和73-98%的ee获得含有手性全碳四元中心茚的衍生物。此外,通过DFT计算,提出了反应机理和合理的立体控制。
Chiral Aluminum Complex Controls Enantioselective Nickel‐Catalyzed Synthesis of Indenes: C−CN Bond Activation
Tao Zhang Yu‐Xin Luan Su‐Juan Zheng Qian Peng* Mengchun Ye*
Angew. Chem. Int. Ed. ASAP, DOI:10.1002/anie.202001142

正文
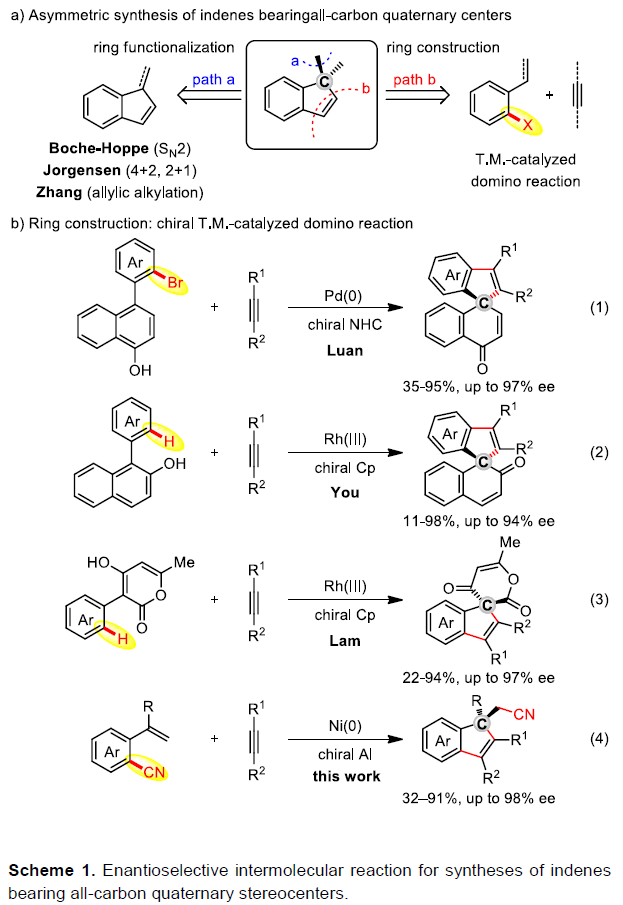
具有全碳四元立体中心的茚类化合物,作为生物活性分子和有机材料中的重要结构单元。大量文献已报道通过外消旋合成法来制备此类结构,但以对映选择性的方式合成此类结构却很少被研究,由于具有刚性环以及位阻碳中心从而使不对称控制变得非常困难。茚环的直接官能化反应,作为典型的分子间不对称方法(Scheme 1a-a),包括丁基锂参与的SN2反应、有机催化剂促进的[4+2]和[2+1]环化反应以及手性膦促使的烯丙基烷基化反应。鉴于茚环的制备相对困难性,通过过渡金属催化的多米诺反应似乎更为有效,因为此策略可以同时将芳烃和炔烃引入到茚环中,同时获得全碳四元产物(Scheme 1a-b)。尽管大量文献已经报道了类似的不对称多米诺反应,用于合成多种带有非全碳四元中心的茚衍生物,但是构建手性全碳四元中心一直是一个巨大的挑战。直到2015年,Luan课题组以C-Br键为多米诺反应的起点,通过Pd催化下的去芳化反应,以对映选择性的方式获得高达97%ee的螺二氢茚衍生物(Scheme 1b-1)。而You课题组,将芳基C-H键代替C-Br键作为多米诺反应的起点,以相似的条件获得高达94%ee的螺二茚衍生物(Scheme 1b-2)。同年,Lam课题组还报道了通过多米诺反应获得一系列螺环茚衍生物,ee高达97%(Scheme 1b-3),其中芳基C-H键仍作为反应的起点,但烯醇化过程用来代替去芳化步骤。尽管已取得了巨大的进展,但对从简单易得的芳烃合成带有全碳四元立体中心的茚类衍生物仍然需求很高。本文中,作者首次报道了一种新的多米诺反应,该工艺以C-C键为起点,以32-91%的收率和高达98%的ee合成多种手性非螺环茚衍生物(Scheme 1b-4)。该策略不仅消除了C-H多米诺反应中对导向基团和其他氧化剂的要求,同时允许一步完成三个C-C键的形成(原子经济性),从而获得含烷基腈基的茚衍生物。
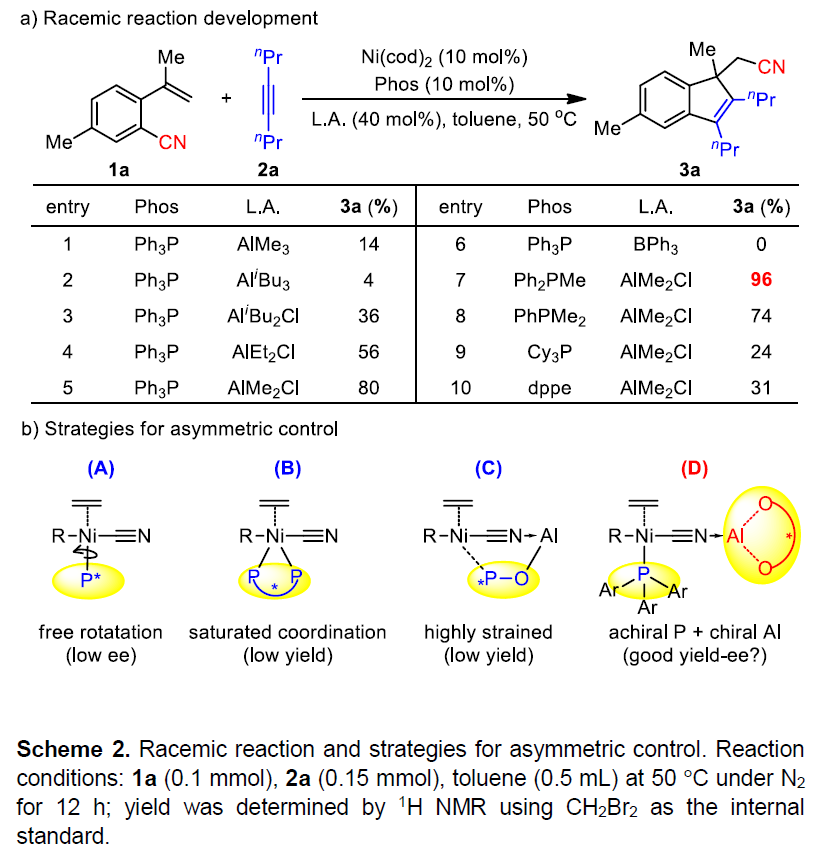
通过过渡金属催化C-CN键活化已被广泛研究,可实现π-不饱和化合物(如炔烃,二烯和烯烃)的羰基化反应,其中路易斯酸助催化剂与膦配体对于反应至关重要。因此,作者选择芳基腈1a和二炔基2a作为模型底物,对膦配体和路易斯酸进行了广泛的筛选(Scheme 2a)。当使用PPh3作为配体时,AlMe2Cl作为最佳路易斯酸,以80%的产率获得所需的环化产物3a(entries 1-6)。随后,以AlMe2Cl作为路易斯酸,筛选了不同的膦配体(entries 7-10)。反应结果表明,Ph2PMe的产率高达96%,而三烷基膦和双膦收率均明显偏低。在获得上述的最佳外消旋条件后,作者探索了几种不对称的策略(Scheme 2b, A to C)。但是,令人失望的是,在所有反应条件下均获得低产量或低ee的产物,由于Ni-P键的自由旋转导致单膦的ee偏低(A),而镍的饱和配位阻碍了烯烃与催化剂配位从而导致双齿膦的低反应性(B)。本课题组先前成功的仲氧化膦(SPO)配体也不理想(C),因为线性Ni-C≡N-Al物种不能同时与SPO配体中P-和O-末端配位,否则易变环。基于上述分析,作者设想一种新形不对称策略:使用非手性单膦和手性铝(D),由于单膦可确保良好的反应性,而手性铝可实现良好的对映选择性。
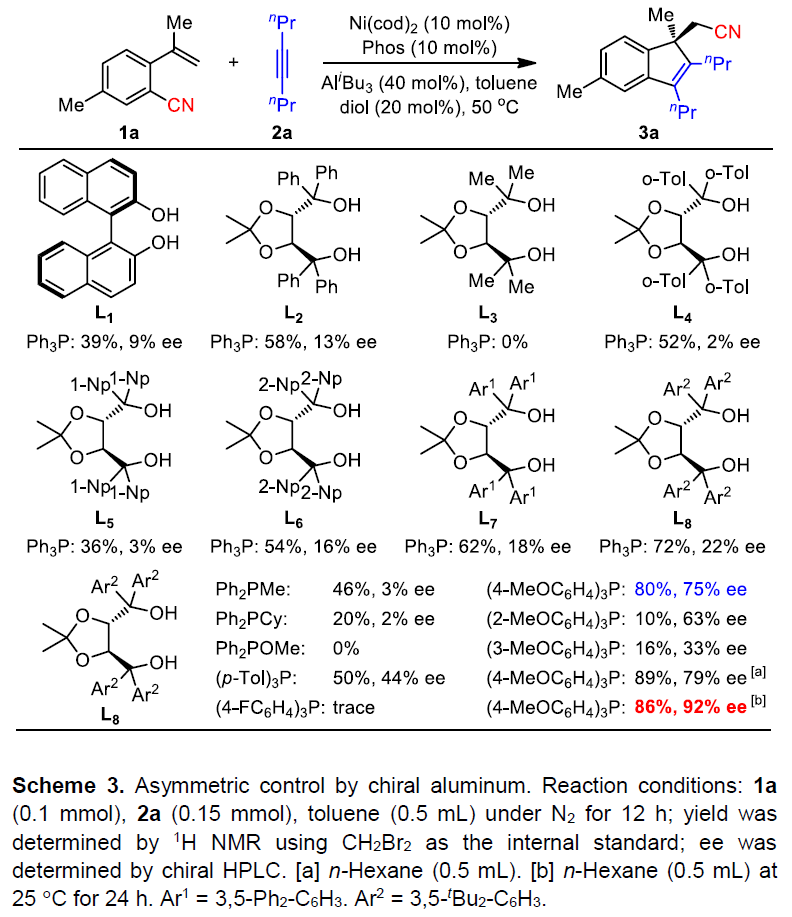
基于上述的研究,作者开始尝试各类非手性单膦和手性铝的相关研究(Scheme 3)。当使用Ph3P(10 mol%)作为非手性膦时,AliBu3(20 mol%)和(R)-BINOL(20 mol%)1:1混合,无法获得产物,可能是由于形成了酸性较低的二烷氧基铝化合物,而将AliBu3(20 mol%)和(R)-BINOL(40 mol%)1:2混合,以39%的收率和9%ee获得所需的产物3a。因此,作者开始筛选了多种二甲醇衍生物(L1–L8),结果表明,L8作为最佳配体,获得产率为72%,ee为22%的3a。为了进一步提高对映选择性,作者在L8-AliBu3的基础上研究了各种单膦,烷基或杂原子取代的膦、芳基环对位上带有吸电子取代基(F)的三芳基膦均阻碍了反应。然而,在芳环上带有给电子基团(Me、MeO)的三芳基膦,可将ee大大提高,如 (4-MeOC6H4)3P可获得产率为80%和75%ee的产物3a。值得注意的是,(2-MeOC6H4)3P也可以获得较高的ee,但收率偏低。此外,较大的位阻也阻碍了随后烯烃的插入。最终,通过使用非极性正己烷和较低的温度(25℃),可获得最佳结果(86%收率,92%ee)。
在获得上述最佳反应条件后,作者开始对底物芳基腈1和炔烃2进行了相关的扩展(Scheme 4)。当芳基腈的芳环上带有中性基团(3a–3d)或给电子基团(3e–3g)时,均可获得相应的产物(收率为62-90%,ee为86-95%),但含有吸电子基团时(如F、CF3、Cl、CO2Me等),ee显着降低。尽管在烯烃的2位上含有tBu和芳基等大基团阻碍了反应,但Et(3l)和线性烷基链(3m和3n)可获得高达98%ee的产物。此外,其它对称烷基炔烃(3o、3p和3q)也可进行反应获得相应产物(收率为70-78%,ee为93-95%)。值得注意的是,当使用非对称的芳基-烷基炔烃时,可获得单个异构体(3r),可能是由于在过渡状态下形成了较大的π-域导致。
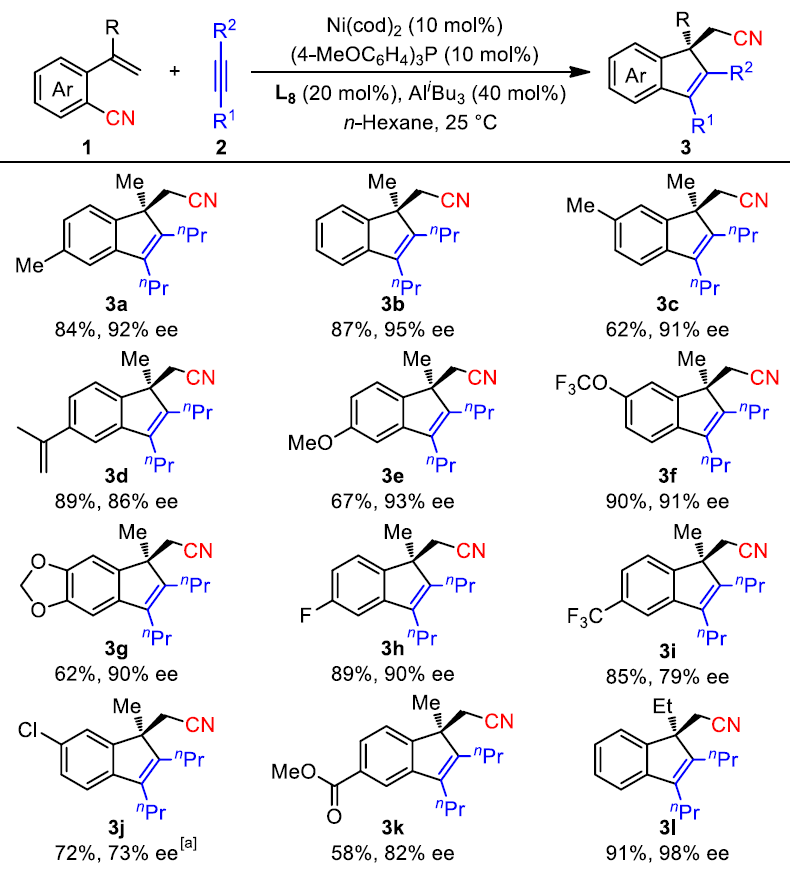

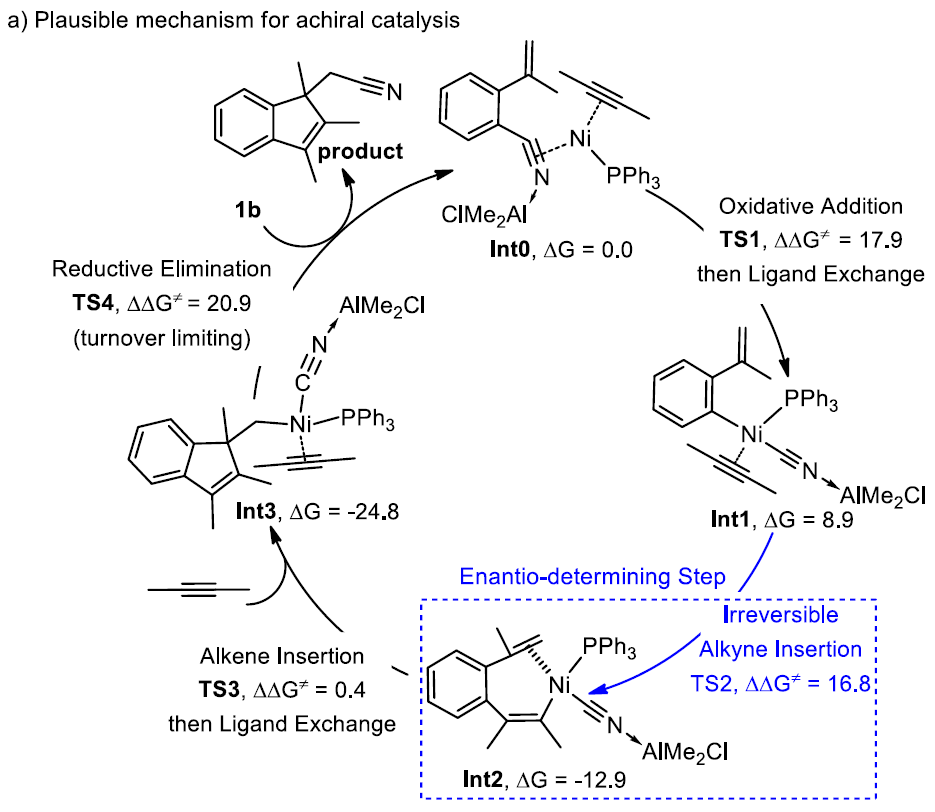
为了进一步了解反应的机理,作者使用2-(丙-1-烯-2-基)苄腈(1b)和丁-2-炔的反应进行相关DFT计算(Scheme 5a)。首先,腈和炔与镍中心配位形成的起始中间体Int0。随后,C-CN键与Ni(0)的进行氧化加成,再与Al配位,经配体交换形成四配位的Ni(II)中间体Int1。当芳基和CN处于反式位置时有利于炔烃的插入,导致中间Int2与前手性烯烃与Ni(II)中心配位。紧接着,烯烃的插入非常容易(能量屏障仅为0.4 kcal/mol),这表明进一步的配体交换和烯烃解离将被限制。最后,经还原消除Int3,获得产物并再生了Ni(0)催化剂,同时该步还决定了反应速率。
总结
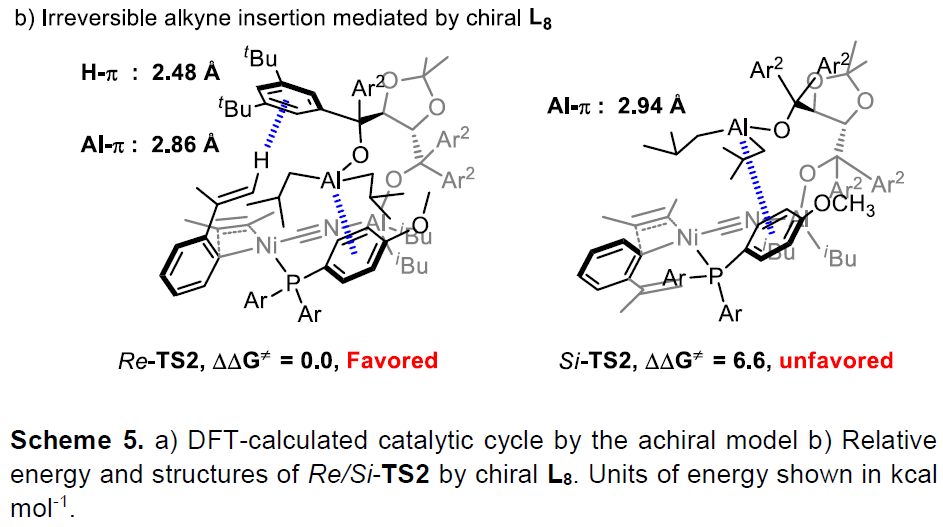
南开大学叶萌春教授和彭谦教授课题组开发了一种镍催化手性铝控制的对映选择性的多米诺反应,合成多种含有全碳四元立体中心茚的衍生物(收率32-91%,ee值73-98%)。该反应使用芳基腈和炔作为起始原料,通过C-CN键活化一步构建了三个C-C新键,体现了原子经济性的特点。此外,该方法将廉价的非手性膦与手性铝相结合,极大地促进了反应的活性和选择性。DFT计算揭示了反应机理,通过手性铝配体与Ni催化反应中心的远程螯合,实现炔烃对映选择性插入控制。
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!




















No comments yet.