

投稿作者:林康杰
Chemistry界有一句很著名的话:Chem is try。在很多研究者的印象中,尤其是刚入行不久的新手,会有一种刻板印象,认为化学研究尤其是有机化学很大程度上是试出来的。而事实上,化学领域有一个分支叫理论和计算化学。其中,很大一部分研究者是用量化的手段研究化学问题。但是,由于化学问题的复杂度,用量化手段往往花费时间较长且很难算复杂度较大的体系,目前还很难做到指导具体的化学实验。而化学信息学作为计算化学里的一个分支,随着近年来机器学习的蓬勃发展,越来越受到部分实验化学家的重视。化学信息学一直都在致力于用建模的方式来定量的研究化学问题。其中,建模的手段很多都和现在的机器学习算法高度相关。随着越来越多化学数据库的公开,data-driven的化学性质和合成路线预测也受到广泛关注。最近,来自UIUC的Scott E. Denmark教授在Science发布了一套基于分子描述符和实验数据驱动的高选择性手性催化剂预测工作流程(workflow),且用来指导合成了高选择性的手性催化剂。
“Prediction of higher-selectivity catalysts by computer-driven workflow
Andrew F. Zahrt*, Jeremy J. Henle*, Brennan T. Rose, Yang Wang, William T. Darrow, Scott E. Denmark
,Science 2019, ASAPDOI: 10.1126/science.aau5631”
——workflow原理介绍——
这个workflow包含以下部分,如图1所示:(i)构建一个可合成的有特定骨架的催化剂的虚拟库。(ii)计算每个分子的化学描述符,创建包含虚拟库的化学空间。(iii)选择代表该化学空间中催化剂的子集。这个子集就称为universal training set (UTS)。这个分类与具体的反应和机理无关,是基于一种名为Kennard-Stone的抽样算法。(iv)收集train set数据。(v)应用机器学习方法来生成预测每个反应的对映选择性模型。模型的评估需要UTS之外的test set。验证过的模型可以用来选择给定反应的最佳催化剂。此时,预测得到的催化剂数据可以和train set重新组合,一起来训练数据来制作更强大的模型。然后可以迭代地重复该过程,直到达到最佳选择性。
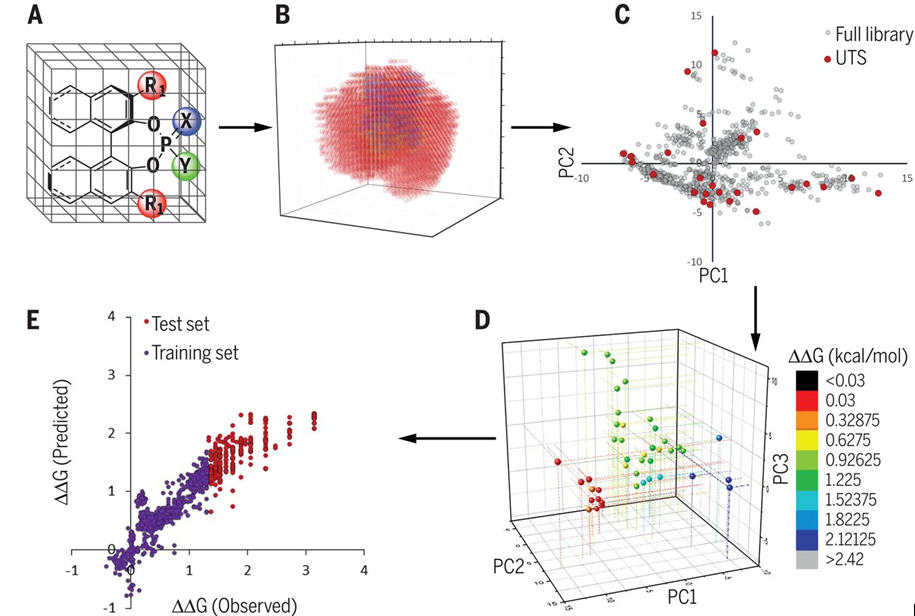
图1. (A) 大型候选催化剂库的生成. (B) 化学描述符的计算. (C) 选择有代表性的UTS数据点. (D) 预测结果平均绝对误差. (E)预测高选择性的反应。
为了开发这个工作流程,作者选择了BINOL(1,1′-联-2-萘酚)衍生的手性磷酸作为催化剂骨架。因为这个系列的化合物易合成且易多样化的添加取代基。作者选择了两个不同BINOL骨架,然后将403个易合成的取代基加入到骨架里边,构建出了含有2*403=806个催化剂的虚拟库。
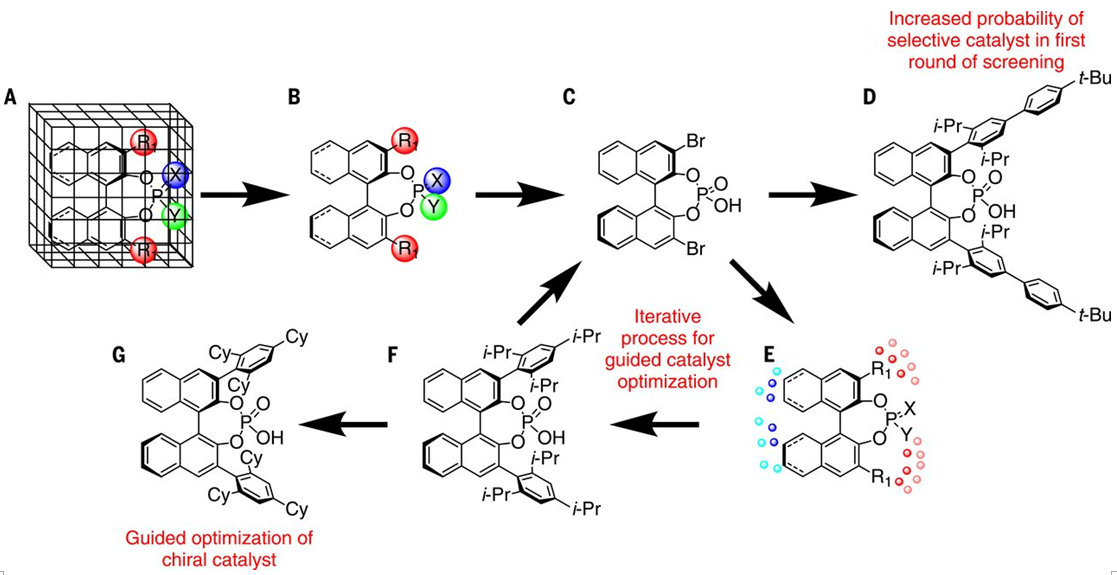
图2.化学信息学指导工作流程示意图。
——空间平均占有率描述符——
作者首先要找到合适的描述符来数字化分子的空间和电子结构。他们先是尝试了常用的0D,1D,2D和3D描述符,但结果都不理想。分析可能是由于只考虑了每种催化剂的一个构象。因为催化剂在溶液中构象都是可变的,很难找到一个准确描述催化效果尤其是不对称催化所需的构象。因此,必须开发出一套新的包含整个conformerensembles的信息的描述符,可用于任何不对称催化剂骨架。作者发明的新的描述符称为平均空间占有率(ASO),主要用来评价空间信息,将conformer ensembles的信息转换为特定位置的数值形式。 ASO的计算流程如图3A所示。首先,计算计算库里的每种催化剂的conformer分布;然后,将每个分子的构象对齐并单独放置在相同的网格中。如果格点是在一个原子的范德华半径范围内,它会被赋值为1;否则就赋值为0。重复对n个构象操作,完成时每个网格点都有一个从0到n的累积值。然后通过除以n归一化,使得所有网格点的值介于0和1之间。如图3B所示,这些值构成了分子结构的空间描述符。红色网格点标记远离催化剂的区域(ASO值为0.000~0.125),而蓝色网格点表示ASO值在0.875到1.000之间。因为催化剂是骨架对齐的,所以相应的ASO接近1。绿色和黄色的格点则代表了相应取代基的旋转位置。这些描述符区分不同类别的催化剂的能力如图3D所示。不同的催化剂类别在化学空间中有不同的分布。
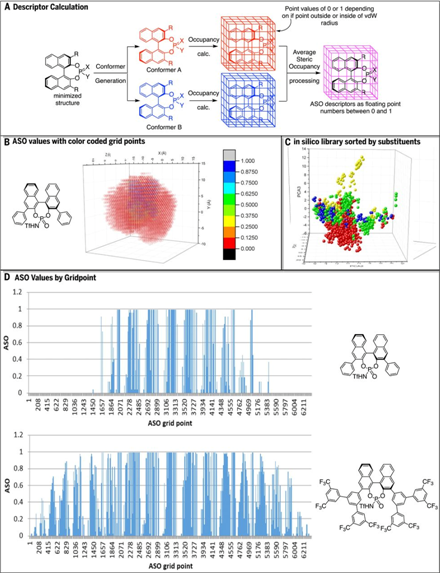
图3. ASO描述符的构建思路。
而电子描述符则来源于取代基施加的对季铵离子的静电势图的扰动,这些基于取代基的电子描述符与空间描述符ASO相结合,则有了表示催化剂的电子和空间结构的特征。每个催化剂的总特征达到了16384个,随后会删除总特征里方差为零的feature。
——UTS子集的构建——
为了选择一个化合物在高维空间的代表性子集,必须对其高维空间进行降维。作者采用主成分分析(PCA)降维,保留了使方差最大化的新维度。随后,作者使用Kennard-Stone算法(从样本集中选择代表性样本的算法)抽样选出代表性子集。这个抽样方法保证来自均匀区域的催化剂特征空间的采样。所选催化剂的子集构成UTS,然后可以用来优化能被该催化剂类型催化的反应。具体过程如图4所示。从UTS中选出了24个代表性的手性磷酸催化剂。同时为了检验UTS的预测结果,另外单独测试19个UTS之外的虚拟库催化剂。外部数据集的选择标准是基于化学家的直觉和所选择催化剂的可合成性。
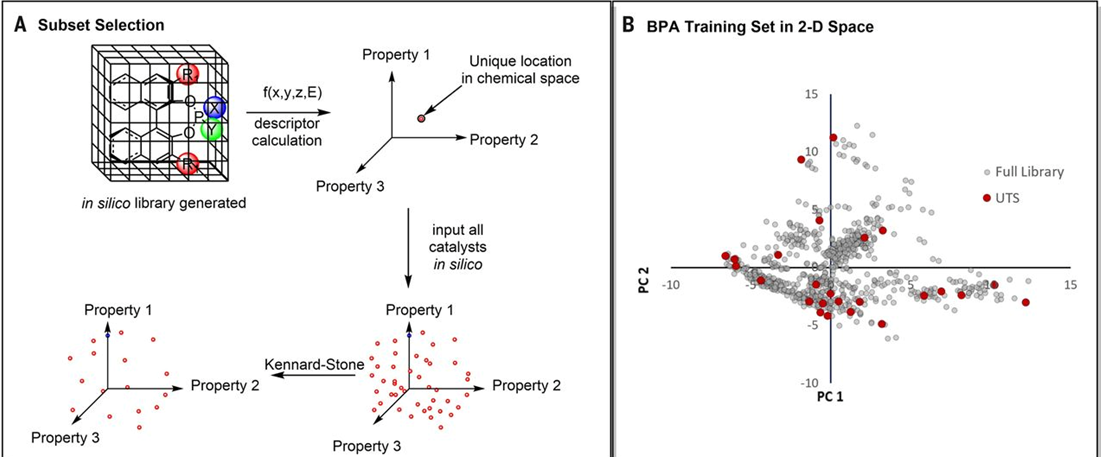
图4. UTS子集的构建及选取。
——方法检验——
为了验证ASO和训练集选择方法,作者采用手性磷酸催化的硫醇与芳基亚胺的不对称加成作为模板反应(因为该反应产率较高且易重复)。分别进行25个反应,计算反应物/产物/催化剂的ASO和电子描述符,得到(24 + 19) *25 = 43*25=1075个数据点。随机选取600个作为train set,其余475作为test set。使用机器学习里常用的支持向量机(SVM)进行建模,实现了对该反应催化剂的对映选择性的准确预测,如图5所示,ΔΔG(反应对映选择性的过渡态能垒差)的平均绝对偏差(MAD)为0.152kcal/mol,优于现有的基于量化的预测结果。当只用随机抽取的384个数据点作为train set,模型的MAD也只低于0.236kcal/mol。
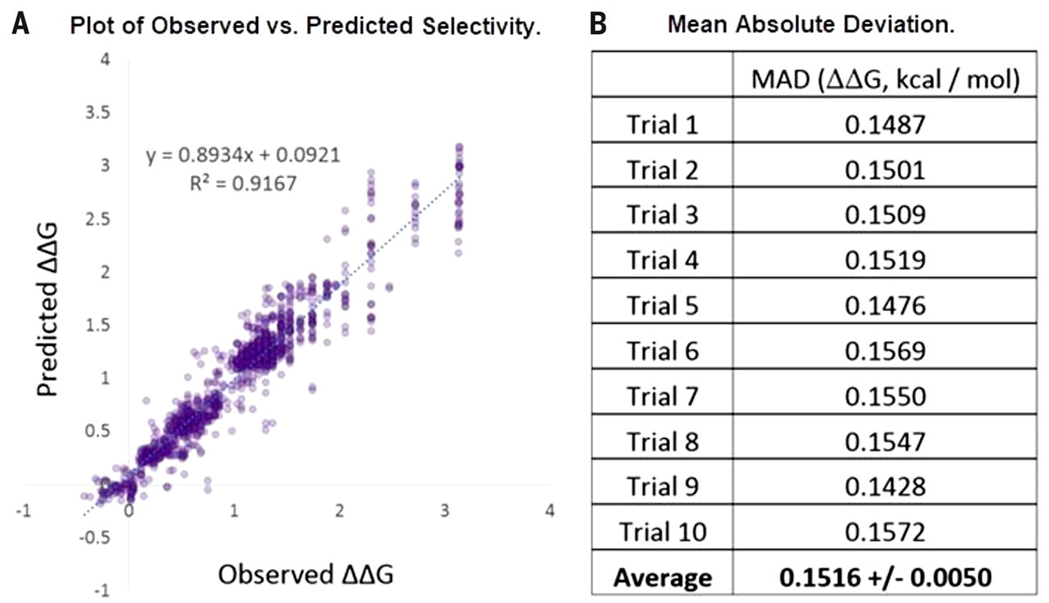
图5.预测结果。
作者为了更好模拟现实情况,只采用较差的低于80%ee选择性的数据点作为train set(718个),来预测高于80%ee选择性的催化剂及反应物组合(357个)。通过测试几个常用的机器学习算法,发现使用前馈神经网络方法得到的结果最好(MAD=0.33 kcal/mol),而且还预测出了选择性最好的52号和另外两个次好的53和54号催化剂。

图6.预测结果。
——结语——
总的来说,作者利用空间描述符ASO和电子描述符作为输入,使用SVM模型成功预测出选择性最好的手性催化剂。并能指导相应的催化剂设计。但是目前该模型还仅限于预测特定类型的催化反应,很难泛化到其它的反应类型。不过,可以肯定的一点是,建立有效的分子描述符来准确描述有机物的空间结构和电子性质,将会对有机化学的研究产生重大影响。尤其是随着高通量筛选的普及,大量优质数据的获取将会变得更容易,也会对化学研究的范式产生深远影响。
参考文献:
Andrew F. Z. et al.“Prediction of higher-selectivity catalysts by computer-driven workflow and machine learning”.Science 363, 247 (2019). DOI: 10.1126/science.aau5631
本文版权属于 Chem-Station化学空间, 欢迎点击按钮分享,未经许可,谢绝转载!


















No comments yet.